扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们

理想的色谱谱带是呈现正态分布(Normal Distribution)[也被称为高斯分布(Gaussian Distribution)]但是现实情况完美的对称峰形在实际的色谱图中是很少见的。绝大多数的峰都会存在不同程度的拖尾,而且随着色谱柱的使用一般拖尾现象会加剧。然而也有一些其它的可能原因造成峰的拖尾(或者前延),因此随时追踪峰形以估计什么时候实际问题会发生是非常有必要的。
基本概念
有两种最常见的峰形评价参数:
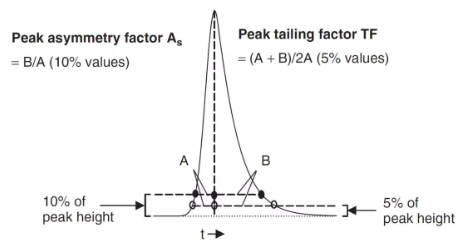
拖尾因子(Tailing Factor,TF):这是在制药行业中使用的峰形评价参数。如上图右边部分,其定义为:
TF = (A + B)/2A
其中A和B为5%峰高处的左右半峰宽;
对称因子(Asymmetry Factor,As):这是制药行业以外的其他行业使用的峰形评价参数,如上图左边部分。其定义为:
As = B/A
其中A和B为10%峰高出的左右半峰宽。
TF和As的近似关系为:
As ≈ 1 + 1.5(TF - 1)
大部分情况下,As的值比TF大,如:
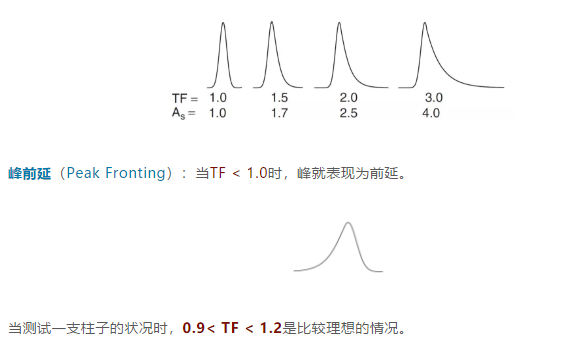
下面将逐一分析HPLC峰发生拖尾、前延和裂分时的原因和解决办法。
峰拖尾
HPLC色谱峰出现拖尾的可能原因有很多,比较常见的有:
1. HPLC体系中存在过大的死体积。这些过大的死体积往往存在于进样模块和检测模块,从而导致一部分的分析物在这些空间里滞留过长的时间(超过正常的保留时间分布带)而表现为拖尾。这种拖尾是对于每一个色谱峰都存在的,因此当出现每个色谱峰都拖尾时,有可能就是这个原因。
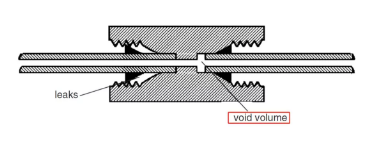
应对方法:更换更短、内径更小的管路;检查部件的连接是否紧密;检查保护柱或者在线过滤器的安装是否合适(这个尤为常见);换到表现正常的仪器排查下是否是色谱柱的原因等。
2. 在HPLC柱中存在两种或以上的吸附位点,这两个位点有不同的平衡等温线和不同的传质动力学速率。简单来说就是色谱柱固定相与分析物作用的非均一性,固定相表面存在大量低能量吸附位点,这种吸附位点与分析物的作用是非选择性的(nonselective),包括色散或者简单的极性作用,同时也存在少量的高能吸附位点,这种吸附位点与分析物的作用是有选择性(selective)或者特异性(specific)的,包括氢键作用、离子交换作用和螯合作用等(如下图)。
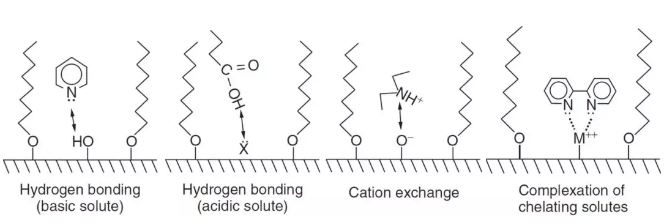
应对方法:最根本的是分析化合物与色谱柱可能存在的各种作用,然后通过改变流动相条件(如添加剂,pH值等)或者针对性地更换色谱柱来减少拖尾。这个问题太大,以后有时间会单独写一篇文章来讨论。
3. 色谱柱过载。当样品进样量过大,使色谱柱出现过载时,色谱峰也会表现出拖尾,这种拖尾峰呈现“鱼鳍”状,非常好判断(如下图)。我们在分析一些化合物,特别是带电荷的化合物时,当保留在固定相上时会因为静电排斥而大大降低其载样量。
应对方法:可以通过降低进样量观察峰形是否改善来确定是否是过载导致的峰拖尾。对于由于带电荷的化合物,可以通过调节pH值(比如把化合物调至中性状态)或者加入离子对试剂来改善峰形(可参见RP-HPLC中的离子对试剂(二):几类重要的IPRs)。
4. 色谱柱柱头污染或筛板部分堵塞。这样导致分析物分子中的一部分滞留时间增加从而导致拖尾。
应对方法:这种情况一般表现为色谱图的所有色谱峰都出现拖尾,可以通过更换同款色谱柱来排查或者反接色谱柱(如果确定色谱柱可以反接)观察峰形是否改善来判断。如果确定是柱头污染或筛板部分堵塞而导致的拖尾,而且还想抢救一下的话,可以参见C18柱:我觉得我还能抢救一下!
峰前延
色谱峰前延出现的频率一般比拖尾少得多,可能的原因有:
1. 色谱柱中出现空洞或柱床塌陷。这常见于填装工艺较差的色谱柱(现在比较少见),或者柱子长期超过其耐受pH范围使用导致硅胶溶解、空洞形成。
应对方法:抱歉!这种情况几乎无法抢救,只能更换色谱柱。对于价格比较昂贵的制备柱,可能可以通过专业的修复来一定程度恢复分离效果。
2. 样品稀释剂洗脱能力较强而导致的“溶剂效应”。进样后一部分分析物分子扩散到流动相氛围,而另一部分然后在稀释剂氛围中,由于稀释剂洗脱能力更强、移动更快,会导致色谱峰出现前延甚至裂分(下面会提到),溶剂效应一般对出峰时间靠前的分析物影响较大。
应对方法:a. 在不影响分析物溶解的前提下,尽量用(初始)流动相溶解样品或者用弱洗脱强度溶剂稀释样品以降低溶剂效应;b. 在不影响灵敏度的前提下降低进样体积;c. 在不影响分离的情况下,有文献建议在柱前安装一段较粗的管路(最近也有公司开发了溶剂效应消除器,可以在网上搜索了解详细信息)。
3. 色谱柱过载。前面提到色谱柱过载时会出现“鱼鳍”峰,但在一定的过载程度上会表现为峰前延。
应对方法:a. 降低进样量;b. 换成载样量高的色谱柱。
4. 柱温过低。对于某些特殊的样品,可能存在不同的互变状态,在柱温太低时由于影响动态平衡,可能会导致色谱峰前延。
应对方法:考察不同的温度对峰形的改善。
峰裂分
色谱峰裂分的部分原因可能和峰拖尾和峰前延一样,当程度不一样时,就可能表现为裂分,如:
1. 色谱柱过载;
2. 色谱柱柱头污染或筛板部分堵塞;
3. 样品稀释剂洗脱能力较强而导致的溶剂效应;如果是这三种原因,应对方法同上。此外,还有可能是:
4. 化合物自身的原因。比如化合物中存在多种互变形式,比如1,3-二羰基类化合物的烯醇互变(如下图)等;或者存在多个离子化位点,如EDTA,当其同时存在不同的离子化状态时,可能导致色谱峰裂分成双重甚至多重峰。
应对方法:考察不同极性溶剂、流动相pH、温度等对峰形的改善情况,也可以针对性地选择特殊的色谱柱固定相,比如键合了带电基团的色谱柱来分析化合物等。由于篇幅有限,后面有机会专门整理一个专题。
总结
HPLC峰形问题中的拖尾、前延和裂分是经常困扰分析人员的问题,而且同一种现象其中的原因往往各不相同,因此问题会呈现不同的复杂程度。本文相对系统地整理了这些问题的可能原因,但由于自身积累和文献整理能力有限,难免会有所遗漏,希望大家可以在留言区踊跃补充。在实际工作中,我们需要不断学习和积累经验,具体问题具体分析,从而快速解决问题。
-END-
参考文献
1. L. R. Snyder, J. J. Kirland, Introduction to Modern Liquid Chromatography 3rd. 2010, John Wiley & Sons, Inc.
2. C. Hawkins, J. Dolan, Understanding Split Peaks. LCGC North America, 2003, 21, 1134.
3. J. C. Giddings, Kinetic Origin of Tailing in Chromatography. Analytical Chemistry, 2000, 35, 1999.
4. J. W. Dolan, Peak Shape Problem. LCGC Europe, 2008, 21.
5. T. Fornstedt, G. Zhong, G. Guichon, Peak Tailing and Mass Transfer Kinetics in Linear Chromatography. Journal of Chromatography A, 1996, 741, 1.
6. T. Fornstedt, G. Zhong, G. Guichon, Peak Tailing and Mass Transfer Kinetics in Nonlinear Chromatography. Journal of Chromatography A, 1996, 742, 55.
文章来源:药视网
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。