扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们

来源
中国现代应用药学 2020 年7月 第37卷第14期
作者
马骏威,刘涓,刘永辉,任连杰
国家药品监督管理局药品审评中心
摘要
目的 阐述强制降解试验研究现状,汇总各强制降解试验基本条件,为药物研发提供借鉴和参考。
方法 汇总实际工作和文献中药物降解基本方法,分析药品注册申报各阶段对降解研究的要求。
结果 现阶段各国监管机构没有提出统一明确的强制降解试验条件,药物的降解条件与药物分子结构及制剂处方相关。
结论 强制降解研究可以帮助确定药物的降解途径并解析降解产物,为药物安全性和毒理学研究提供支持,也为稳定性指示分析方法、药物处方、包装选择和储存条件的开发提供支持。从药物研发早期到后期,强制降解需要不断完善。
关键词
强制降解;药物研发;降解条件;注册申报
_
正文
_
药物的有效性和安全性与药物稳定性密切相关。药物储存期间杂质水平的增加可能会影响活性成分的药理作用,甚至导致不良反应[1]。强制降解是在较短时间内采用剧烈条件强制药物产生一定水平的降解,用于预测实际稳定性放置期间可能存在的降解产物。
1、强制降解研究的目的
强制降解试验也称为破坏性试验,开展强制降解试验的主要目的包括[2]:
①解析降解产物,确定药物的降解途径和机制。药物的降解途径与药物分子结构密切相关,分子结构中存在酯基或者酰胺键在酸碱催化条件下可能发生水解,胺基容易得氧、烯丙基和苄基容易失氢氧化。
②为药物安全性提供支持。当毒理学相关杂质不容易获得时,对一定程度降解样品进行毒理学评价,能够为药物安全性及杂质限度确定提供支持性依据。
③有助于代谢产物研究。有些降解产物同时也是代谢产物,降解试验样品产生的代谢产物可以用于确证研究和分析。
④有助于原料药工艺、制剂处方和工艺的开发,有助于药物盐型和晶型的筛选。当降解杂质具有毒性结构时,可通过变更工艺路线避免杂质产生;也可控制工艺参数确保杂质在可接受水平。不同晶型和盐型药物的强制降解结果可指示其稳定性。⑤开发具有稳定性指示能力的分析方法。稳定性指示方法的定义是经过验证的可以检测原料药和制剂的化学、物理或微生物特性随时间变化的定量分析方法,而且是特定的,使得主成分、降解产物和其他待测成分可以在没有干扰的情况下进行准确测量[1]。
⑥指导药物包装系统开发,确定储存条件。降解试验结果可提示药物的敏感性因素。强制降解试验、影响因素试验、加速试验和长期试验结果共同支持药物包装和储存条件。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
2、强制降解试验条件的选择
2.1 强制降解的基本要求
强制降解条件应尽可能保证药物有一定程度的降解,一般是5%~20%的降解量。
原料药的降解一般会考察其在固体和溶液状态的情况。原料药固体状态一般考察在光照、热和湿热条件的降解情况,选择性进行氧化降解;原料药溶液或混悬液状态一般需进行高温、水解、氧化以及光照等试验。原料药强制降解试验应设置非降解组作为对照组。
制剂的降解条件选择与剂型密切相关。对于固体制剂(如片剂、胶囊等),降解研究一般包括热、湿热、光照和氧化。对于溶液/混悬液制剂(如注射液、口服混悬液),降解研究包括热、水解、氧化和光照。制剂应设置非降解组作为对照组,同时为排除辅料自身降解的干扰,建议选择安慰剂作为强制降解研究的空白对照组[3]。实际工作中某原料药在各降解条件下稳定,但制剂降解明显,提示制剂中辅料可能与原料药发生作用,需继续关注样品长期稳定性数据。
建议选择一批样品进行强制降解研究,如结果不明确,则需要加试几批样品。强制降解试验需考虑受试品的物理性质,采取冷藏和/或密闭等措施,降低样品物理状态变化(如升华、蒸发、熔化)对实验结果的干扰,试验容器应选择惰性容器。强制降解试验应保证样品有相对较大的暴露量。以光照试验为例,原料药固体进行光照试验,药物需均匀分散在容器中,厚度≤3 mm,固体制剂采用单层平铺方式等;例如,索非布韦原料药粉末在培养皿中铺展至约1 mm 厚度,在80 ℃下72 h 进行热降解[4]。对于临床使用有特点的制剂,如输液、皮肤用霜剂等,需结合实际使用进行设计[5]。对于需配成溶液的制剂,建议从低浓度开始直至临床使用浓度,确定不同浓度药液对降解产物的影响。
2.2 强制降解试验条件
药物典型的降解条件包括高温、光照、氧化和水解4 种。不同药物在不同条件下降解程度不同,例如OPSUMIT® (macitentan) 在酸和碱条件下水解产生10 个降解产物,而在氧化、中性水解、热和光条件下基本不降解[6]。原料药降解条件较制剂更为剧烈,例如酸碱水解条件,原料药可能会选择1~2 或12~13 的pH 值,而制剂一般选择制剂目标pH 值附近±2 个pH 单位,主要原因可能是原料药降解研究,重点在于药物的降解途径和机制探索,而制剂的降解研究更关注模拟实际储存及生产的情况。强制降解具体试验条件汇总见表1。
高温降解温度一般在60~105 ℃。水解反应一般在室温下进行,可根据需要升高温度[7]。通常采用HCl 和NaOH(0.1~1 mol·L-1)溶液进行水解。水解结束后,使用合适的酸、碱或缓冲液中和溶液以避免进一步降解。对于难溶于水的药物,可以使用助溶剂或共溶剂进行溶解。应注意,使用的共溶剂或助溶剂应不与药物分子发生作用。如利伐沙班在水中难溶,溶于乙腈制备0.5 mg·mL-1 溶液,因结构中包含酰胺键,其在0.1 mol·L-1 NaOH(3∶1) 60 ℃ 4 h 基本降解完全[8]。
强制光照可考虑选择人用药品注册技术国际协调会议(international council on harmonization,ICH) Q1B 建议光照(总照度1.2×106 lux·h,近紫外能量≥200 Wh·m-2)的2~5 倍强度条件。
药物氧化降解途径主要包括2 种,第一种是经自由基链引发的氧化,许多因素可以加速氧化,如氧浓度(如150 psi 加压氧)、温度、pH、金属离子、光照和其他可能的自由基引发物等。氧气可以使氨苄青霉素(ampicillin,AMP)降解,AMP 和Cu(II)摩尔比为1∶1 时,pH 9.0 下降解最快,这与形成的中间活性氧相关[9]。制剂辅料(如聚维酮、羟丙基纤维素、交联聚乙烯吡络烷酮、聚乙二醇、聚山梨酯等)中的某些杂质,如醛类、金属离子、过氧化物类,可作为氧化反应的引发剂,引发自由基链反应导致制剂发生氧化降解[10]。


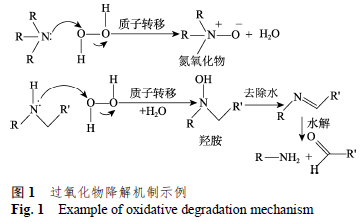
第二种氧化途径是通过有机过氧化物和/或过氧化氢氧化。过氧化物降解机制见图1[2]。许多药物分子的特定基团易于发生氧化降解,例如,药物中的叔胺和仲胺基团会与氢过氧化物发生亲电反应形成N-氧化物和羟胺;硫化物易受电子转移氧化得到砜和亚砜[11]。依达拉奉结构中含有吡唑酮,6% H2O2 条件下室温避光15 min降解即可达到17%[12]。达卡他韦在溶液中,其氨基甲酸酯部分容易在碱性条件下水解,其咪唑基团容易发生碱介导的自氧化,也可在过氧化氢条件下氧化[13]。罗红霉素片原研和自制制剂在3%过氧化氢条件下即剧烈降解[14]。
3、药物不同开发阶段对于强制降解试验的要求
药学开发的进度与药物临床进程密切相关,随着药物研发的深入,对药物降解产物结构解析、降解途径的理解也需要不断深入。尽管在临床Ⅰ、Ⅱ期,各监管机构对强制降解研究未提出明确的要求,但尽早掌握药物的降解情况和水平,有利于药物理解,降低开发风险。
一般认为临床前即可以开始进行强制降解研究,此时可以不进行降解产物的解析,强制降解试验研究主要为了开发有关物质分析方法。国内Ⅰ期或Ⅱ期的临床申报一般不需要报告强制降解研究条件或结果,建议结合影响因素试验对于可能的降解途径进行讨论[15],应说明杂质分析情况,包括杂质来源、结构、限度等,进行简单的杂质分析。
美国FDA 对于强制降解研究也是逐渐深入[16]。FDA 要求鼓励研发早期进行原料药的强制降解试验,临床Ⅱ期应提交有关降解方面的数据更新,以便能够进行安全性评价,开发稳定性指示分析方法;FDA 要求将降解试验的研究结果纳入Ⅲ期临床申报,以证实原料药的内在稳定性、潜在的降解途径及拟用分析方法的能力与适用性、安全性,适当时,应鉴定、定性、定量并报告降解产物。
国内注册上市申报阶段,有关物质研究部分要求进行强制降解试验以支持分析方法的专属性研究,同时需结合影响因素试验和加速试验的结果对降解产物进行分析研究[17]。ICH M4Q 中要求,上市申报阶段需提交强制降解和影响因素试验用于表征分析方法的适用性[18]。
4、讨 论
4.1 影响因素试验、强制降解试验的区别
影响因素试验在国内稳定性指导原则的翻译是stress testing,属于正式稳定性研究的一部分。中国稳定性指南中明确,应考察原料药和制剂对光、湿、热、酸、碱、氧化等的敏感性、主要的降解途径及降解产物,为验证所用分析方法的专属性、确定加速试验的放置条件及选择合适的包装材料提供依据。国内外稳定性指南中关于stresstesting 的试验要求是基本一致的,均提供了建议的试验条件。原料药通常在比加速条件更剧烈的条件考察,高温、高湿试验通常设定5,10,30 d 考察。制剂影响因素试验包括光稳定性以及某些制剂的特定试验,如注射液冻融试验[19]。实际工作中,也建议研究制剂在高湿和高热等条件下的稳定性。但是国内外stress testing 论述中均没有提供酸、碱、氧化降解的明确的试验条件,这可能是因为不同药物条件的选择是不同的,无法统一规定。
影响因素考察结果不强调要达到一定的降解量,试验结果主要用于指示药物对光照、湿、热等因素的敏感性。相比影响因素试验,强制降解试验更侧重于降解目标,即达到一定的降解量。其降解条件可能与影响因素相同,也可能不同。对于影响因素试验条件下药物没有达到理想程度的降解,需要采用更加剧烈或者更加有针对性的方法降解。一定程度的降解样品有助于稳定性指示方法的建立。但并不是要盲目地追求药物的降解,因为过于剧烈的条件有可能引发过度降解或者二级降解,这些降解杂质在实际稳定性储存期间可能不会产生。一些稳定药物在剧烈条件下均不降解,此时可通过加标已知杂质模拟降解样品来指导分析方法的开发。
4.2 仿制药强制降解试验
原研药品上市时,由于缺乏相关信息,需要开展完整的强制降解试验。仿制药研究应先调研已有公开文献或信息,初步评估可能降解产物和降解途径。其次,开发者应充分评估自制产品与原研品的差异,包括原料药的起始物料、合成路线、纯化工艺、结晶条件、晶型、包装容器,根据评估结果,有针对性地确定降解条件。建议对原研和自制产品进行降解研究对比,确定降解途径和杂质谱的区别。对于研究但没有降解产生的情况,FDA 在仿制药申报要求中建议申请人重新进行强制降解试验,以获得足够多的降解产物。如果仍然没有降解产生,需要阐明依据[20]。
4.3 强制降解知识的积累
目前强制降解试验条件的选择主要依靠人的经验。随着大量数据的积累,对于降解产物的共享、降解途径的预测变得越来越重要。辉瑞公司建立了降解数据库,可以方便研发人员通过结构检索已完成鉴定的降解产物,并获得类似结构物质的降解产物信息[21]。也可采用计算机方法来预测降解产物的方法, 例如耶鲁大学化学院Jorgensen Research Group 于1980—1985 年开发的CAMEO(计算机程序),可以预测在给定原料、试剂和条件下有机反应的产物[3]。这些降解数据库及计算机方法的发展与完善,为今后药物强制降解试验的研究提供了支持。
5、展 望
强制降解研究主要用于降解途径和降解产物研究,对于稳定性指示分析方法的开发、药物安全性评估、生产工艺和包装系统的开发都至关重要。无论是仿制药还是新药,降解研究都是药物研发必不可少的重要组成部分。
降解研究并不是目的,降解程度尽量要与实际生产和储存中产生杂质建立一定的相关性。对于在多种条件下仍然不能产生降解的稳定药物,也可以采用加标的方法或者生产过程中粗品作为样品,用于分析方法的开发。目前除了依靠研发经验,也可以采用已有数据库的方法进行强制降解研究。笔者认为通过强制降解研究可以初步掌握药物杂质谱,对药物中可能存在的杂质有初步认识,特别是对于药物中可能存在高毒性化合物越早的发现与认识,如黄曲霉素类杂质、遗传毒性杂质等,能够提示开发者关注工艺的合理性,降低杂质水平,进而降低药物研发安全性和成药性的风险。
参考文献
详见 中国现代应用药学 2020 年7月 第37卷第14期
文章来源:凡默谷
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。