扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 01
01
GMP体系~变更定义
在GMP体系中,变更~是指对已获准上市化学药品在生产、质控、使用条件等多方面提出的涉及来源、方法、控制条件的变化。可以说,为了提高产品质量,以生产出安全、有效、稳定的产品为目的所做的更改都可视为变更。而这些变化是一种主动的预先计划的改变,可能影响到药品安全性、有效性和质量可控性。变更的范围包括原辅料、包装材料、质量标准、检验方法、生产工艺、操作规程、厂房、设施、设备、仪器的变更。

02
GMP体系~变更分类
根据变更的性质、范围和对产品质量潜在的影响程度,以及变更是否能够影响注册、变更时限等,有不同的分类方法,可根据自身实际情况选择适合自身企业的分类方法,变更分类包括但不局限于如下所列:
重大变更、一般变更和微小变更(目前企业应用的最多)
重大变更(需要向药监管理部门提交文件申请批准),是指会对产品质量有显著性影响的变更,主要包括:产品注册标准的变更;内包装材质的变更;储存条件、标签、说明书的变更;关键合成路线、主要原料或溶剂、关键工艺参数、处方成分发生改变;运行方式、构造发生改变的设备变更以及生产厂址的变更等。
一般变更(药政部门备案),是指对产品内在质量(安全性、有效性)存在潜在影响的变更,主要包括:生产、质量的主要设备、设施、仪器的关键部件的更换;关键原辅料的生产合成工艺、质量标准、供应商的改变;清洁方法的改变;包装规格和标签变更;GMP文件的改变等。
微小变更(不需要向药监部门申请批准或备案),是指对产品的安全性和有效性不会产生影响的变更。这类变更只需写进年度质量报告中。主要包括:设备设施的辅助配件的更换;质量标准未改变的原辅料供应商的变更;中间体生产场地的变更等。
涉及注册的变更、不涉及注册的变更
在国内的现行药品生产法律法规中多次涉及到变更的条款,如《药品生产质量管理规范》均明确规定药品生产企业应严格按照注册批准的生产工艺生产;规范中多次提到包括生产工艺规程、操作规程或 SOP 药品储存条件等均应按注册标准进行。
《药品注册管理办法》更是多处提到了注册中对于变更的具体要求,如变更研制新药、生产药品和进口药品已获批准证明文件及其附件中载明事项的,应当提出补充申请;修改药品注册标准、变更药品处方中已有药用要求的辅料、改变影响药品质量的生产工艺等的应报补充申请;改变国内药品生产企业名称、改变国内生产药品有效期、国内药品生产企业内部改变药品生产场地等应报补充申请;变更药品包装标签应报补充申请;等。
03
GMP体系~变更控制
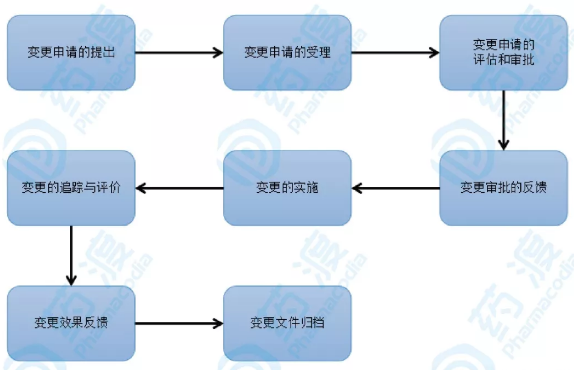
GMP体系中,变更控制是保证任何对验证过的工艺、程序、文件、设备、设施、供应商、物料以及电子软件系统的变更在执行前都经过记录、评估和批准。

变更的主要负责部门,通常是由质量管理部门来协调整个变更控制过程,要承担以下工作:
1、协助发起人对变更进行评估和分析;
2、指定所有和这个变更有关的部门来审核,如果变更需要报批国家药品监督管理部门批准,必须通知相关人员进行申报;
3、将各部门的意见反馈给发起人;
4、监督在变更过程中需要完成的各项工作是否都按照要求彻底完成;
5、对变更进行趋势分析。
对一个已经验证过的系统的变更往往会伴随着无法预知的风险,因此,在一个变更的发起阶段,就应该进行详细的评估来判断这个变更是否必要。这个评估可以从以下方面来进行:
1、是否是药监部门的要求?
2、是否影响到员工的安全?
3、是否是法规符合的要求?
4、是否会提高产品的质量或生产效率?
小结/小感
在刚刚过去的2018,国内制药业所发生一系列的疫苗事件,其中就有很大一部分原因来自生产过程中对于变更的隐瞒,其影响极为恶劣,不但企业自身自食其果,也对整个行业造成了非常大的负面作用!药品,作为特殊的商品,关乎生命,故药品全生命周期的质量控制,自然就极为重要!而由于技术的进步、质量标准的不断提升,生产中相关变更的出现,是非常正常且有意义的事情。虽然GMP追求稳定,而变更的本质不是在挑战稳定,相反,却是为了更为长远的稳定,而我们每个制药人所要做的,就是如何使变更更好的得到控制,更好的为药品的属性服务!
文章来源:GMP验证及信息化
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。