扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 摘 要
摘 要
杂质作为药品的一项关键质量属性,是贯穿于研发始终的一项重要研究内容。杂质谱分析是杂质研究工作的基础,通过全面的杂质谱分析,可指引药品制备工艺的开发和优化、质量控制策略的制定;可使杂质检查工作有的放矢,是建立合理可行检查方法的基础。本文主要是根据笔者几年来的审评工作经验,结合案例对化学合成原料药的杂质谱分析的一般原则、研究思路进行初步的探讨。
任何影响药物纯度的物质统称为杂质,ICHQ3A中对新原料药的杂质定义为:存在于新原料药中,但化学结构与新原料药不同的任何一种成分。药品在临床使用中产生的不良反应除了与药品本身的药理活性有关外,有时与药品中存在的杂质也有很大关系。例如,青霉素等抗生素中的多聚物等高分子杂质是引起过敏的主要原因。杂质作为药物的一项关键质量属性,是研发工作的一项重要研究内容。
按杂质的化学类别和特性,可分为:有机杂质、无机杂质、有机挥发性杂质。杂质谱分析是对药品中各种可能存在的杂质的概貌掌握,通过全面的杂质谱分析,可指引药品制备工艺的开发和优化、质量控制策略的制定;可使杂质检查工作有的放矢,根据不同杂质的特性来针对性的建立检查方法,有助于检查方法的建立和验证。原料药的杂质谱分析对应于CTD格式申报资料的模块“3.2.S.3.2杂质”。本文主要是根据笔者几年来的审评工作经验,结合案例对化学合成原料药的有机杂质谱分析的一般原则、研究思路进行初步的探讨,不涉及检查方法的研究及验证的内容。
1.有机杂质分析的一般考虑
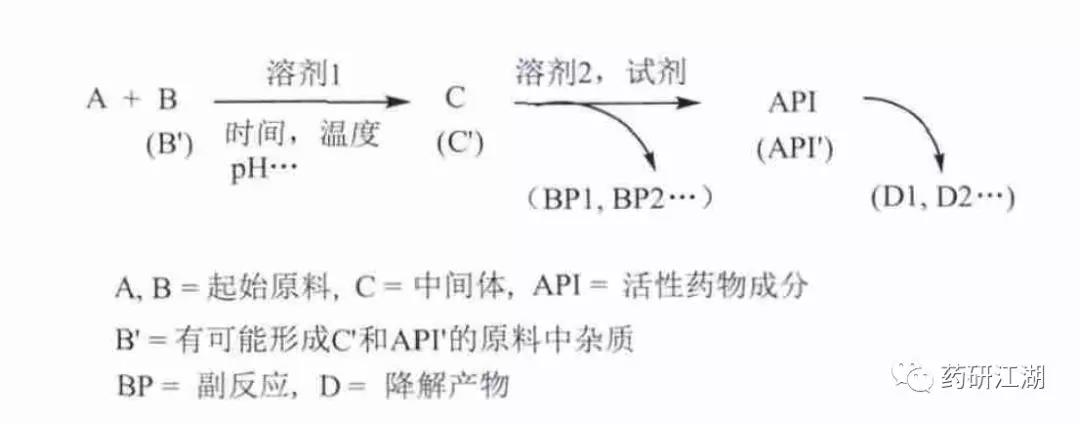
图1有机杂质潜在来源示意图
图1是一个化学合成原料药的工艺示意图,是以A和B为起始原料,经中间体C制备最终产品(API),根据此反应通式,有机杂质的潜在来源主要包括以下几个方面:①合成原料(A、B)及中间体(C);②合成原料引入的杂质(如B′),以及合成原料引入杂质的后续反应产物,如B′的后续反应产物C′、API′;③副反应产物,如BP1、BP2等等,如果副反应产物可随主成分一同参与后续的反应,还需关注其后续反应产物;④原料药的降解产物,如D1、D2等等;⑤反应中使用的试剂、配位体、催化剂等等。由于这类杂质的化学结构一般与活性成分类似或具有渊源关系,通常也称为有关物质。除了降解产物外,其他这4类有机杂质都与制备工艺有关,也称为工艺杂质。
审评工作中发现,申请人进行杂质谱分析时多存在两种极端的问题,一种极端是杂质分析过于简单,例如,工艺杂质仅关注合成原料和中间体;另一种极端是杂质分析过于繁杂,例如,将有机合成的各种理论可能的副反应杂质均简单罗列,但不做进一步的分析讨论、杂质检查。ICHQ3A中明确要求:申请人应对原料药在合成、精制和储存过程中最可能(mostlikely)产生的那些实际存在的和潜在的杂质进行综述分析。因此,杂质谱分析的对象应是那些最可能产生的工艺杂质和降解产物,而不是工艺中使用的所有物料、试剂的简单罗列,也不是理论上所有可能副反应的简单排列组合。合理的杂质谱分析应建立在对合成所涉及的化学反应、由原料引入的杂质及可能的降解产物进行科学合理评估的基础之上。不仅要关注实际检出的杂质,还需要对潜在杂质存在的可能性进行科学评估,以更有效地指导有关物质检查方法的筛选和建立。
2.工艺杂质的分析
工艺杂质的分析应基于工艺开发过程中知识和数据的积累,对制备工艺以及所涉及化学反应机理要有深入的理解,要注意分析工艺中杂质的形成、去向(是否可随主成分一同进行后续的化学反应)及清除情况。总之,掌握的信息越丰富,就越容易评估哪些是在终产品中最可能存在的杂质。
对于工艺中使用的合成原料、中间体以及试剂、配位体、催化剂等等,相对比较简单,申请人基本都能关注到此类潜在工艺杂质的残留问题。下面主要对其他几种工艺杂质进行讨论。
2.1原料引入的杂质
建议根据供应商提供的制备工艺,对外购起始原料可能引入的杂质进行全面的分析和检测,注意分析起始原料引入杂质在后续工艺步骤中的去向/清除情况,结合后续中间体中控实测数据的积累,合理制定起始原料引入杂质的质控策略(源头控制,过程控制,或在终产品中继续关注)。
建议重点关注那些可引入后续反应的潜在杂质,通常这类杂质的结构与主成分类似,可随主成分一同进行后续的化学反应,且理化性质也可能与主成分比较接近,后续工艺步骤对其清除能力相对其他杂质来说比较有限,在终产品中残留的可能性也较大,这类杂质也多见用于有关物质检查方法系统适用性的分离度规定。例如,EP8.0收载塞来昔布标准中的杂质A就是合成原料引入的苯环间位甲基异构体杂质(图2)随主成分一同进行后续反应而产生的工艺杂质,因其结构和性质与主成分非常接近,比较难以清除,不仅作为特定杂质规定限度0.4%,还在系统适用性中规定主成分峰与杂质A峰的分离度不低于1.8。
图2塞来昔布和EP杂质A的结构图
2.2副反应杂质
建议根据工艺开发过程中掌握的工艺认知、对所涉及化学反应机理的理解以及数据的积累,对各步骤可能产生的副反应杂质进行合理分析,并跟踪其在后续工艺步骤中的去向/清除情况,根据多批次跟踪数据的积累,合理制定各工艺副产物杂质的质控策略。同样,建议重点关注与主成分结构类似、可引入后续反应的副产物杂质。
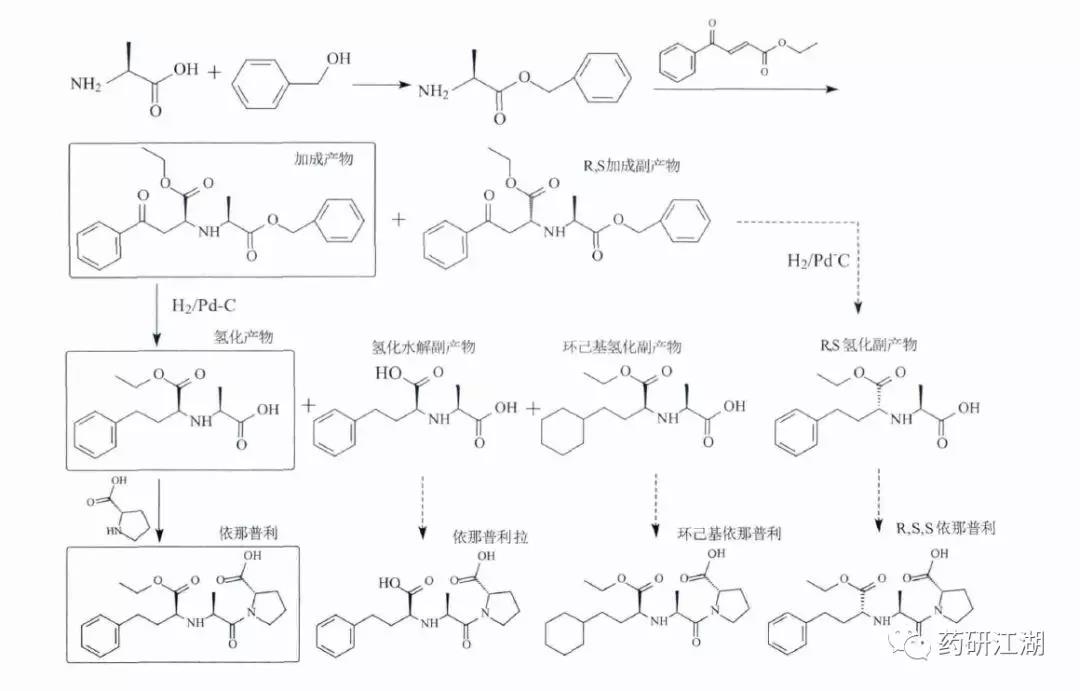
图3依那普利工艺副产物分析图
以马来酸依那普利为例进行说明,简略合成路线见图3,以L-丙氨酸为原料,经酯化、加成、催化氢化、成酰胺等多步反应制备马来酸依那普利原料药。其结构中含有3个手性中心,第2个手性中心是在加成反应中引入,通过溶解性的差异可分离、去除该步骤产生的非对映异构体杂质RS加成副产物,但是少量残留的该副产物杂质可继续参与后续的氢化、酰化反应,最终生成终产品的RSS异构体杂质。此外,在Pd/C催化氢化步骤中,可能发生乙酯水解、苯环氢化的副反应,这些副反应杂质均可参与下一步的酰化反应,最终生成工艺杂质依那普利拉、环己基依那普利。以上工艺副产物杂质产生的可能性较高,还需要终产品质量标准的控制,EP8.0收载的马来酸依那普利原料药标准中RSS异构体杂质(EP杂质A,限度1.0%)、依那普利拉(EP杂质C,限度0.3%)、环己基依那普利(EP杂质H,限度0.3%)均作为特定杂质进行了质控,系统适用性中还规定了RSS异构体杂质(EP杂质A)与主峰的分离度。
3.降解杂质的分析
建议可通过结构特征的分析以及试验的手段来研究潜在的降解途径和降解产物,稳定性试验、强制降解试验是常用的试验手段。相对于稳定性试验,强制降解试验可在较短的时间内获得大量的有益信息,因此在早期研发阶段,强制降解试验是研究潜在降解途径和降解产物的一种有效手段,此外,它还可帮助建立专属性的有关物质检测方法,为制剂的处方、工艺、包材等开发工作提供有益信息。
ICHQ1A中说明强制降解试验的内容包括热、湿、氧化、光照、水解等;试验样品可采用固体、溶液/混悬液的状态;对于试验条件,因不同产品的稳定性不同,指南中仅笼统的说明,强制降解通常是在比加速试验更剧烈的条件下进行,例如,高温试验条件一般是高于加速试验温度10℃以上(如50℃、60℃等)。在审评工作中经常看到申请人对试验条件缺乏必要的筛选考察,要么条件过于剧烈,产生大量无意义的次级降解,要么条件过于温和,未能体现应有的降解,无法达到强制降解试验的研究目的。建议根据原料药本身的物理化学稳定性筛选考察合适的强制降解试验条件,通常主成分降解10%左右即可(也有研究者推荐主成分降解5%~20%)。对于比较稳定的药物,也没有必要采用过于剧烈的试验条件使其必须达到某种程度的降解。
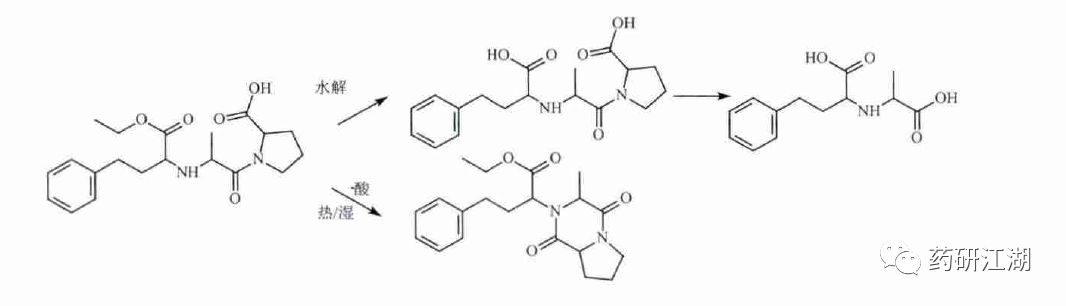
仍以依那普利为例进行说明(图4),通过溶液以及固体状态下的降解试验,依那普利的主要降解途径是水解降解,在酸碱条件下首先是酯键的水解,如果条件剧烈,还可能进一步产生酰胺键的水解产物;在酸水解、固体湿热条件下还可见形成内酰胺环合的降解产物。
4.其他
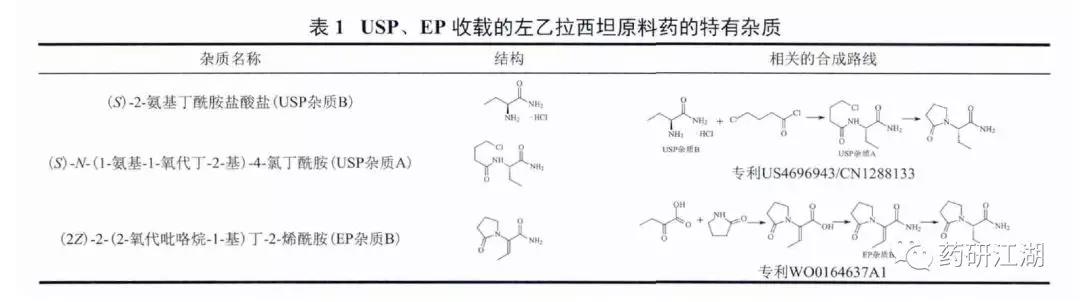
药典等文献也是杂质谱分析的重要参考,可参考文献中报道的同品种或相同结构类型药物的杂质信息。但需要注意的是,由于合成路线可能不同,不建议简单套用文献报道的工艺杂质,需要结合自拟的合成工艺分析是否可能产生与文献报道一致的杂质。例如,因合成工艺不同,USP38、EP8.0的左乙拉西坦原料药标准中收载了不同的特定工艺杂质,(S)-2-氨基丁酰胺盐酸盐(USP杂质B)和(S)-N-(1-氨基-1-氧代丁-2-基)-4-氯丁酰胺(USP杂质A)是USP收载的特有杂质,(2Z)-2-(2-氧代吡咯烷-1-基)丁-2-烯酰胺(EP杂质B)是EP收载的特有杂质,分别产生于不同的合成路线(表1);毒性杂质2-羟基吡啶(EP杂质C)虽然在USP38、EP8.0中均有收载,也建议结合自拟制备工艺(是否使用该合成试剂)考虑是否需要作为特定杂质进行研究。
5.小结
杂质研究是贯穿于药品研发始终的一项重要内容,杂质谱分析是杂质研究工作的基础,基于杂质谱分析的杂质控制是“质量源于设计”的基本理念在杂质研究与控制中的一种具体实践。通过本文的讨论,希望给研究者一些有益的启示,从杂质谱分析入手确立科学的杂质研究思路。
文章来源: CROU制药在线
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。