扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们

Annex 1, Manufacture of Sterile Medicinal Products
附录1,无菌药品生产
Figure 6 and Table 6 identify the 14 most frequent requirements cited in Annex 1. The most frequent citation addresses precautions to avoid contamination and the second addresses environmental monitoring in the areas where aseptic operations are performed. The third most frequent requirement cited is associated with setting appropriate alert and action limits for the results of environmental monitoring. Five requirements are tied for 10th place, so they are all included.
图6和表6列出了附录1的14个最常见的法规要求引用。最常见的引用涉及避免污染的预防措施,第2个引用涉及无菌操作区域的环境监测。第3个最常提到的要求是为环境监测结果设置适当的警戒和行动限。有5项法规要求由于并列第10都包括在内。
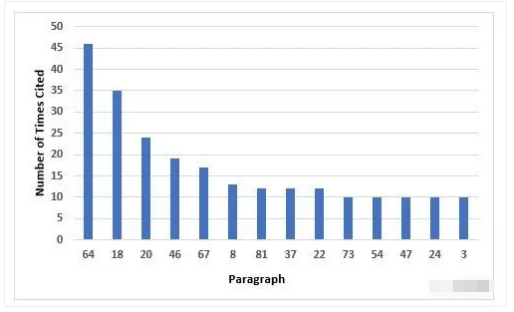
Figure 6: Top 10 deficiencies citing Annex 1
附录1的Top10缺陷引用
Table 6: Top 10 Deficiencies Citing Annex 1
附录1的Top10缺陷引用
|
Paragraph |
# |
Short Text |
|
64 |
46 |
Precautions to minimize contamination should be taken during all processing stages including the stages before sterilisation. 应在灭菌前的所有工艺步骤采取预防措施以尽量减少污染。 |
|
18 |
35 |
Where aseptic operations are performed monitoring should be frequent using methods such as settle plates, volumetric air and surface sampling (e.g. swabs and contact plates). Sampling methods used in operation should not interfere with zone protection… 在进行无菌操作时,应经常使用沉降菌、浮游菌和表面菌(如擦拭和接触板)等方法进行监测。操作中使用的取样方法不应干扰区域保护。 |
|
20 |
24 |
Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring. If these limits are exceeded operating procedures should prescribe corrective action. 尘埃粒子及微生物监测结果应设定适当的警戒及行动限。如果超过这些限度,操作规程应规定纠正措施。 |
|
46 |
19 |
In clean areas, all exposed surfaces should be smooth, impervious and unbroken in order to minimize the shedding or accumulation of particles or micro-organisms and to permit the repeated application of cleaning agents, and disinfectants where used. 在洁净区,所有外露的表面应光滑、不透水和不破损,以减少粒子或微生物的脱落或积聚,并允许在使用时反复使用清洁剂和消毒剂。 |
|
67 |
17 |
The process simulation test should imitate as closely as possible the routine aseptic manufacturing process and include all the critical subsequent manufacturing steps… 工艺模拟试验应尽可能地模拟常规无菌制造过程,并包括所有关键的后续制造步骤。 |
|
8 |
13 |
Clean rooms and clean air devices should be routinely monitored in operation and the monitoring locations based on a formal risk analysis study and the results obtained during the classification of rooms and/or clean air devices. 洁净房间和结晶空气装置应进行日常动态监测,监测点应基于正式的风险分析研究和在对房间和/或洁净空气装置进行级别确认时获得的结果时获得的结果来确定。 |
|
81 |
12 |
Components, containers, equipment and any other article required in a clean area where aseptic work takes place should be sterilised and passed into the area through double-ended sterilisers sealed into the wall, or by a procedure which achieves the same objective of not introducing contamination… 在进行无菌操作的洁净区,部件、容器、设备和其他任何需要的物品都应进行灭菌,并通过密封在墙上的双扉灭菌器进入洁净区,或通过一种不产生污染的程序进入洁净区。 |
|
37 |
12 |
All personnel (including those concerned with cleaning and maintenance) employed in such areas should receive regular training in disciplines relevant to the correct manufacture of sterile products… 在这些区域工作的所有人员(包括与清洁和维护有关的人员)都应定期接受与正确生产无菌产品有关的培训。 |
|
22 |
12 |
The transfer of materials into and out of the unit is one of the greatest potential sources of contamination… 物料进出转移的环节是最大的潜在污染源之一。 |
|
73 |
10 |
Activities in clean areas and especially when aseptic operations are in progress should be kept to a minimum and movement of personnel should be controlled and methodical, to avoid excessive shedding of particles and organisms due to over-vigorous activity… 在洁净区的活动,特别是在进行无菌操作时,应保持在最低限度,人员的移动应得到控制和并有条理,以避免过度活跃的活动导致粒子和有机体的过度脱落。 |
|
54 |
10 |
It should be demonstrated that air-flow patterns do not present a contamination risk, e.g. care should be taken to ensure that air flows do not distribute particles from a particle generating person, operation or machine to a zone of higher product risk. 应证明气流模式不构成污染风险,例如,应注意确保气流不会将来自产生粒子的人员、操作或机器的粒子散布到产品风险较高的区域。 |
|
47 |
10 |
To reduce accumulation of dust and to facilitate cleaning there should be no uncleanable recesses and a minimum of projecting ledges, shelves, cupboards and equipment… 为了减少灰尘的积累和便于清洁,不应有不干净的凹槽,应尽少存在突出的壁架、架子、橱柜和设备…… |
|
24 |
10 |
Isolators should be introduced only after appropriate validation… 隔离器应该在适当的验证之后引入… |
|
3 |
10 |
Clean areas for the manufacture of sterile products are classified according to the required characteristics of the environment… 用于生产无菌产品的洁净区按所需的环境特性进行分类。 |
Chapter 6, Quality Control
第6章,质量控制
Figure 7 and Table 7 identify the most frequently cited requirements in Chapter 6. The two most frequently cited requirements are the need to adequately control laboratory components and reagents and the need to trend data and investigate OOT (out of trend) and OOS (out of specification) events. Again, we find the importance of investigating such events similar to those identified in deficiencies cited in Chapter 5 and Chapter 1.
图7和表7确定了第6章中最经常引用的法规要求。两个最常被引用的法规要求是充分控制实验室组件和试剂的要求,以及趋势数据和OOT、OOS事件调查的要求。再次,我们发现调查类似于第5章和第1章中所提到的缺陷的事件的重要性。
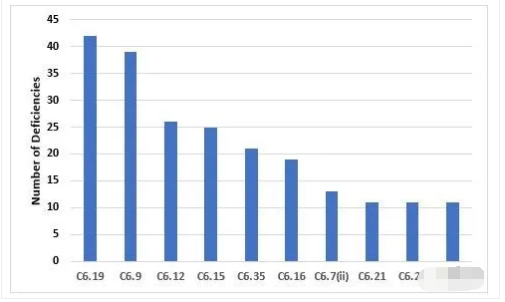
Figure 7: Top 10 deficiencies citing Chapter 6
第6章的Top10缺陷引用
Table 7: Top 10 Deficiencies Citing Chapter 6
第6章的Top10缺陷引用
|
Paragraph |
# |
Short Text |
|
6.19 |
42 |
Special attention should be given to the quality of laboratory reagents, solutions, glassware, reference standards, and culture media… 应特别注意实验室试剂、溶液、玻璃器皿、参考标准品和培养基的质量。 |
|
6.9 |
39 |
Some kinds of data (e.g., tests results, yields, environmental controls) should be recorded in a manner permitting trend evaluation. Any out of trend or out of specification data should be addressed and subject to investigation. 某些数据(如试验结果、收率、环境控制)应以允许趋势分析的方式记录。任何不符合趋势或不符合标准的数据都应加以处理并接受调查。 |
|
6.12 |
26 |
Samples should be representative of the batch of materials or products from which they are taken… 样品应能代表所取批次的物料或产品…… |
|
6.15 |
25 |
Testing methods should be validated…
分析方法应得到验证... |
|
6.35 |
21 |
Out of specification or significant atypical trends should be investigated. Any confirmed out of specification result, or significant negative trend, affecting product batches released on the market should be reported to the relevant competent authorities… 应调查不符合标准或显著的非典型趋势。任何影响产品放行到市场上的确认的不合格结果或显著的负面趋势,应向相关主管部门报告… |
|
6.16 |
19 |
The results obtained should be recorded. Results of parameters identified as quality attribute or as critical should be trended and checked to make sure that they are consistent with each other. Any calculations should be critically examined. 获得的结果应加以记录。确定为质量属性或关键参数的结果应进行趋势分析和检查,以确保它们彼此一致。任何计算都应严格审查。 |
|
6.7(ii) |
13 |
Procedures describing sampling, testing, records (including test worksheets and/or laboratory notebooks), recording, and verifying; 描述取样、测试、记录(包括测试工作表和/或实验室记录本)、记录方式和验证的程序; |
|
6.21 |
11 |
Laboratory reagents, solutions, reference standards, and culture media should be marked with the preparation and opening date and the signature of the person who prepared them… 实验室试剂、溶液、参考标准品和培养基应标明配制和开启日期,并由配制人员签字…… |
|
6.27 |
11 |
The purpose of the ongoing stability programme is to monitor the product over its shelf life and to determine that the product remains, and can be expected to remain, within specifications under the labelled storage conditions. 持续稳定性方案的目的是在有效期内监测产品,并确定产品在标识储存条件下可预期的保持符合质量标准。 |
|
6.5 |
11 |
Control laboratory premises and equipment should meet the general and specific requirements for Quality Control areas given in Chapter 3… 控制实验室的设施和设备应符合第3章对质量控制领域的一般和具体要求。 |
Chapter 8, Complaints and Product Recall
第八章,投诉和召回
Figure 8 and Table 8 identify the top 10 deficiencies that cite Chapter 8. By far the most frequent citation addresses the need to periodically challenge and determine the effectiveness of the recall process. Generally, this exercise is expected to be conducted annually. The next two most frequent deficiencies address the need to implement appropriate corrective and preventive actions in response to quality defects and to report qualify defects to the health authorities when this may result in a product recall.
图8和表8列出了第8章的10大缺陷引用。到目前为止,最常见的引用涉及到定期挑战和确定召回程序有效性的需要。一般来说,这项工作预期每年进行一次。接下来两个最常见的缺陷是针对质量缺陷采取适当的纠正和预防措施的需要,以及在可能导致产品召回时向卫生当局报告确认缺陷。
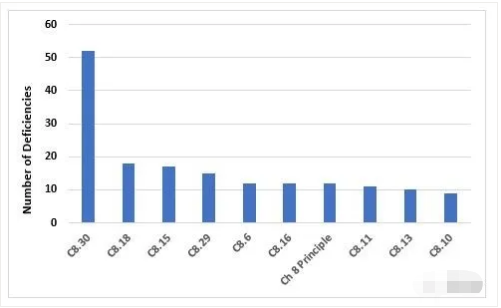
Figure 8: Top 10 deficiencies citing Chapter 8
第8章的Top10缺陷引用
Table 8: Top 10 Deficiencies Citing Chapter 8
第8章的Top10缺陷引用
|
Paragraph |
# |
Short Text |
|
8.30 |
52 |
The effectiveness of the arrangements in place for recalls should be periodically evaluated to confirm that they remain robust and fit for use… 应对召回安排的有效性进行定期评估,以确保其保持稳健并适合使用…… |
|
8.18 |
18 |
Appropriate CAPAs should be identified and taken in response to a quality defect. The effectiveness of such actions should be monitored and assessed. 应针对质量缺陷识别并采取适当的CAPA。应监测和评价这些措施的有效性。 |
|
8.15 |
17 |
Quality defects should be reported in a timely manner by the manufacturer to the marketing authorisation holder/sponsor and all concerned Competent Authorities in cases where the quality defect may result in the recall of the product or in an abnormal restriction in the supply of the product. 在出现可能导致产品召回或产品供应受到异常限制的质量缺陷时,制造商应及时向上市许可持有人/代理人及所有有关主管部门报告质量缺陷。 |
|
8.29 |
15 |
The progress of the recall process should be recorded until closure and a final report issued, including a reconciliation between the delivered and recovered quantities of the concerned products/batches. 应记录召回程序的进展情况,直至完成并发出最后报告,包括有关产品/批次的已交付数量与回收数量之间的平衡。 |
|
8.6 |
12 |
Special attention should be given to establishing whether a complaint or suspected quality defect relates to falsification. 应特别注意投诉或怀疑的质量缺陷是否与造假有关 |
|
8.16 |
12 |
An appropriate level of root cause analysis work should be applied during the investigation of quality defects… 在质量缺陷调查过程中,应使用适当水平的根本原因分析工作…… |
|
Principle |
12 |
In order to protect public and animal health, a system and appropriate procedures should be in place to record, assess, investigate, and review complaints, including potential quality defects, and if necessary, to effectively and promptly recall medicinal products for human or veterinary use and investigational medicinal products from the distribution network… 为了保护公众和动物健康,应该建立系统和适当的程序记录、评估、调查、和审查投诉,包括潜在的质量缺陷,如有必要,有效和及时从分销网络召回人用或兽用药品和临床实验药品… |
|
8.11 |
11 |
If a quality defect is discovered or suspected in a batch, consideration should be given to checking other batches and, in some cases, other products, in order to determine whether they are also affected… 如果在一个批次中发现或怀疑有质量缺陷,应考虑检查其他批次,在某些情况下,检查其他产品,以确定它们是否也受到影响…… |
|
8.13 |
10 |
The decisions that are made during and following quality defect investigations should reflect the level of risk that is presented by the quality defect as well as the seriousness of any non-compliance with respect to the requirements of the marketing authorisation/product specification file or GMP… 在质量缺陷调查期间和之后所做的决定应反映质量缺陷所带来的风险水平,以及任何不符合上市许可/产品标准文件或GMP要求的严重程度。 |
|
8.10 |
9 |
When a quality defect investigation is initiated, procedures should be in place to address at least the following: … 当质量缺陷调查开始时,程序应至少解决以下问题:… |
Chapter 7, Outsourced Activities
第七章,外包活动
Figure 9 and Table 9 show the deficiencies that cite Chapter 7. Among the two most frequent citations is the need for a Quality Agreement and the content of that agreement with regard to the outsourced activity.
图9和表9显示了第7章的缺陷引用。两个最常被引用的是关于外包活动的质量协议和协议内容的法规要求。
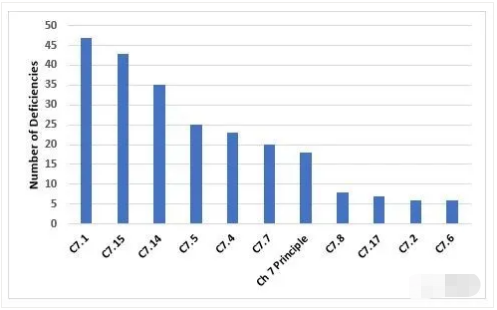
Figure 9: Top 10 deficiencies citing Chapter 7
第7章的Top10缺陷引用
Table 9: Top 10 deficiencies citing Chapter 7
第7章的Top10缺陷引用
|
Paragraph |
# |
Short Text |
|
7.1 |
47 |
There should be a written Contract covering the outsourced activities, the products or operations to which they are related, and any technical arrangements made in connection with it. 应当有一份书面合同,包括外包活动、与之相关的产品或操作,以及与之相关的任何技术安排。 |
|
7.15 |
43 |
The Contract should describe clearly who undertakes each step of the outsourced activity, e.g., knowledge management, technology transfer, supply chain, subcontracting, quality and purchasing of materials, testing and releasing materials, undertaking production, and quality controls (including in-process controls, sampling and analysis). 合同应明确谁承担外包活动的每一步,如: 知识管理、技术转移、供应链、分包、物料质量和采购、物料测试和发放、生产承接、质量控制(包括过程控制、取样和分析)。 |
|
7.14 |
35 |
A Contract should be drawn up between the Contract Giver and the Contract Acceptor which specifies their respective responsibilities and communication processes relating to the outsourced activities. 在外包活动中,委托方与受托方之间应订立合同,明确各自的职责和沟通过程。 |
|
7.5 |
25 |
Prior to outsourcing activities, the Contract Giver is responsible for assessing the legality, suitability and the competence of the Contract Acceptor to carry out successfully the outsourced activities… 在外包活动之前,委托方负责评估受托方成功实施外包活动的合法性、适宜性和能力。 |
|
7.4 |
23 |
The pharmaceutical quality system of the Contract Giver should include the control and review of any outsourced activities… 委托方的药品质量体系应包括对任何外包活动的控制和审查…… |
|
7.7 |
20 |
The Contract Giver should monitor and review the performance of the Contract Acceptor and the identification and implementation of any needed improvement. 委托方应监督和审查受托方的表现,并确定和实施任何需要的改进。 |
|
Principle |
18 |
Any activity covered by the GMP Guide that is outsourced should be appropriately defined, agreed and controlled in order to avoid misunderstandings which could result in a product or operation of unsatisfactory quality… 任何由GMP指南所涵盖的外包活动都应该得到适当的定义、同意和控制,以避免可能导致产品或操作质量不合格的误解…… |
|
7.8 |
8 |
The Contract Giver should be responsible for reviewing and assessing the records and the results related to the outsourced activities… 委托方应负责审查和评估与外包活动相关的记录和结果…… |
|
7.17 |
7 |
The Contract should permit the Contract Giver to audit outsourced activities, performed by the Contract Acceptor or his mutually agreed subcontractors 合同应允许委托方审计由受托方或双方同意的分包商进行的外包活 |
|
7.2 |
6 |
All arrangements for the outsourced activities including any proposed changes in technical or other arrangements should be in accordance with regulations in force, and the Marketing Authorisation for the product concerned, where applicable. 所有外包活动的安排,包括任何拟议的技术或其他安排的改变,均须符合现行法规,以及有关产品的上市许可(如适用)。 |
Annex 11, Computerized Systems
附录11,计算机化系统
The 10 most frequent inspection deficiencies that identify requirements in Annex 11 are shown in Figure 10 and Table 10. The first most frequent deficiency addresses the application of risk management throughout the life cycle of computerized systems. The second most frequent deficiency addresses the failure to ensure integrity of that data and that it is routinely backed up.
附录11中要求的10个最常见的检查缺陷见图10和表10。第1个最常见的缺陷是在整个计算机化系统生命周期中应用风险管理。第2个最常见的不足之处是未能确保数据的完整性和数据的常规备份。
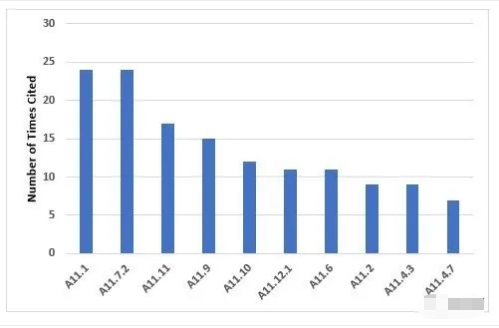
Figure 10: Top 10 deficiencies citing Annex 11
附录11的Top10缺陷引用
Table 10: Top 10 Deficiencies Citing Annex 11
附录11的Top10缺陷引用
|
Paragraph |
# |
Short Text |
|
11.1 |
24 |
Risk management should be applied throughout the life cycle of the computerized system taking into account patient safety, data integrity and product quality… 风险管理应贯穿于计算机化系统的整个生命周期,并考虑到病人的患者安全、数据的完整性和产品质量…… |
|
11.7.2 |
24 |
Regular backups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically. 应定期备份所有相关数据。备份数据的完整性和准确性以及恢复数据的能力应该在验证期间进行检查,并定期进行监控。 |
|
11.11 |
17 |
Computerized systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security and validation status reports… 计算机化系统应定期进行评估,以确认其处于有效状态并符合GMP要求。这些评估应适当包括当前功能范围、偏差记录、事件、问题、升级历史、性能、可靠性、安全性和验证状态报告…… |
|
11.9 |
15 |
Consideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated "audit trail") … 在进行风险评估的基础上,应考虑在系统中创建与GMP相关的所有更改和删除的记录(系统生成的“审计追踪”)…… |
|
A1.10 |
12 |
Any changes to a computerized system including system configurations should only be made in a controlled manner in accordance with a defined procedure. 对计算机化系统的任何更改,包括系统配置,都应按照规定的程序,以受控的方式进行。 |
|
11.12.1 |
11 |
Physical and/or logical controls should be in place to restrict access to computerized system to authorised persons… 应设置物理及/或逻辑控制,以限制有权限的人使用电脑系统… |
|
11.6 |
11 |
For critical data entered manually, there should be an additional check on the accuracy of the data… 对于手动输入的关键数据,应该对数据的准确性进行额外检查…… |
|
11.2 |
9 |
There should be close cooperation between all relevant personnel such as Process Owner, System Owner, Qualified Persons, and IT… 所有相关人员,如过程所有者、系统所有者、QP和IT人员之间应密切合作。 |
|
11.4.3 |
9 |
An up to date listing of all relevant systems and their GMP functionality (inventory) should be available. 应提供所有相关系统及其GMP功能(inventory)的最新清单。 |
|
11.4.7 |
7 |
Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and er
文章来源:允咨GMP制药技术
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。
|