扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 注射剂是指可直接注入人体的给药剂型,包括溶液剂、注射用无菌粉末、注射用浓溶液,以及注射用微球、纳米乳和脂质体等特殊注射剂。由于注射剂是直接注入人体发挥药效,因此相对于口服制剂而言,也是风险比较大的一种剂型。据2016 年度《国家药品不良反应监测年度报告》(CFDA 颁布) 显示,2016 年药品不良反应/ 事件报告中静脉注射给药占59.7%,其他注射给药( 如肌内注射、皮下注射等) 占3.4%,口服给药占33.7%,其他给药途径( 如外用、贴剂等) 占3.2% [1]。目前,因受国内药品销售政策、医院用药习惯等因素的影响,在同一品种存在多种剂型可选的情况下,国内临床偏向于选择注射剂。同时,我国医药行业也正处于产业升级转型时期。正是基于这样的时代背景,2017 年10 月8 日中共中央办公厅国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械》。
注射剂是指可直接注入人体的给药剂型,包括溶液剂、注射用无菌粉末、注射用浓溶液,以及注射用微球、纳米乳和脂质体等特殊注射剂。由于注射剂是直接注入人体发挥药效,因此相对于口服制剂而言,也是风险比较大的一种剂型。据2016 年度《国家药品不良反应监测年度报告》(CFDA 颁布) 显示,2016 年药品不良反应/ 事件报告中静脉注射给药占59.7%,其他注射给药( 如肌内注射、皮下注射等) 占3.4%,口服给药占33.7%,其他给药途径( 如外用、贴剂等) 占3.2% [1]。目前,因受国内药品销售政策、医院用药习惯等因素的影响,在同一品种存在多种剂型可选的情况下,国内临床偏向于选择注射剂。同时,我国医药行业也正处于产业升级转型时期。正是基于这样的时代背景,2017 年10 月8 日中共中央办公厅国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械》。
一.国内注射剂仿制药现状
我国普通仿制药注射剂一般经药学研究审评通过后可直接批准上市,由于我国注射剂仿制药长期以来一直“仿标准”而不是“仿品种”,即只重视与原研的质量标准中指标( 如pH、含量等常规指标)的对比,而未对处方工艺进行认真剖析,因此常常随意更改产品处方、剂型,对产品处方的合理性、生产技术核心等重视不够,造成临床应用中不良反应发生率高于原研制剂,甚至随意更改注射剂生产工艺,如2006 年的“欣弗”事件。目前,国内化学药注射剂常见问题如下。
1.1立题合理性不足
立题合理性问题主要突出表现在改规格、改剂型、改盐基注射剂的品种(“三改”品种) 上,《技术要求》中明确指出:改规格品种需要根据说明书用量论证其科学性、合理性和必要性;改剂型和改盐基品种则需要有明确的临床优势。
1.2 处方工艺与原研不一致
国内注射剂仿制药常见随意更改辅料种类和用量,特别是对于一些重要的功能性辅料的改变,则应该考虑其安全性和有效性。如注射用伏立康唑原研制剂( 威凡) 采用磺丁基-β- 环糊精(SBE-β-CD)进行增溶,而国内产品处方不尽相同,如采用专用溶剂( 丙二醇和乙醇混合溶液) 进行溶解,或采用羟丙基-β- 环糊精(HP-β-CD) 进行增溶[3]。现研究表明[4],SBE-β-CD 对伏立康唑的增溶效果要远优于HP-β-CD,相同辅料用量的情况下,SBE-β-CD对伏立康唑的增溶效果至少为HP-β-CD 的2 倍。
1.3质量控制需要提升
历史上,我国对仿制药质量对比研究不够重视,常以仿制标准如进口注册标准或中国药典(ChP) 为主要目标,因此结果常为质量标准一致,但质量不一致[5]。例如维生素C 注射液收载于ChP 2015 第二部中,其标准中未制定有关物质检查项。孙春艳等对国内维生素C 注射液进行了质量评价,结果表明68 批样品中有6 批杂质总量超过1.0%,建议需要进一步完善质量标准,增加有关物质检查项[6]。
1.4稳定性较差或贮藏条件与原研不一致
稳定性试验是确定药品贮藏条件重要依据之一。我国许多早期仿制的注射剂品种未与原研制剂进行全面的质量对比,并依据稳定性研究结果确定贮藏条件,上市后暴露出很多药品存在质量问题。利福霉素钠及其注射液的仿制均在2005 年以前,原研制剂(Lepeti) 说明书规定的贮藏条件为“2 ~8 ℃冷藏”,同时英国药典(BP) 和欧洲药典(EP)对原料药的贮藏要求均为“2 ~ 8 ℃冷藏”,但我国局颁标准中原料药及注射用的贮藏条件均为“在凉暗处保存”( 不超过20 ℃ ),导致国内生产的利福霉素钠注射液有关物质远远高于原研制剂[7]。
1.5包材质量不符合要求
注射剂是质量风险较高的剂型之一,其包材的好坏可以直接影响到制剂的质量,关系到用药的安全性。玻璃是注射剂常见的包装材料之一,基于成本等影响的考虑,国内90%以上的安瓿采用低硼硅玻璃[8],玻璃输液瓶则多采用钠钙玻璃材质,但是对于一些偏碱或偏酸的药物而言,这类材质的包材常带来一定安全隐患。如碳酸氢钠注射液,目前主要采用药用玻璃( 钠钙玻璃和低硼硅玻璃) 包装,塑包和软袋包装仅约占15%,此类注射剂长期放置易出现玻璃脱片、白点或形成雾状颗粒等问题[9]。
二.国内外技术法规要求
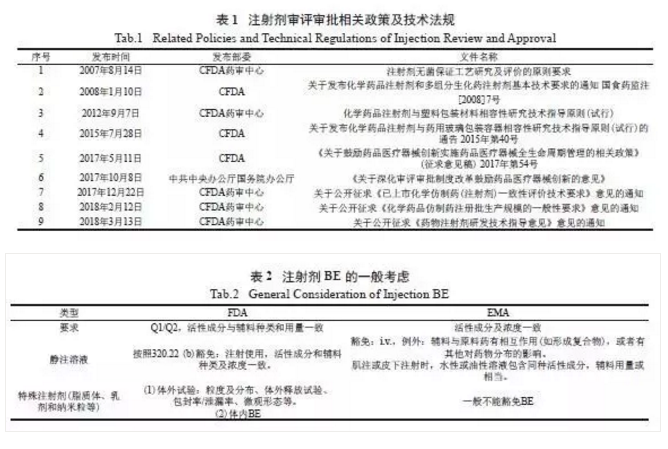
自2007 年我国逐步开始严格注射剂的审评审批,并陆续发布了相关研究技术要求( 表1)。2008年国家食品药品监督管理总局(CFDA) 正式颁布了《化学药品注射剂基本技术要求( 试行)》,对化学药品注射剂的研发提出了明确的技术要求。
三.注射剂一致性评价药学研究要点
2006 年美国FDA 率先提出了“质量源于设计”(Quality by Design,QbD) 的理念,迅速在制药行业内得到广泛认同。自2013 年开始,FDA 要求仿制药的开发和生产均要采用QbD 理念。QbD的核心是基于参比制剂的质量概况(Quality Target Product Profile,QTPP) 和关键质量属性(Critical Quality Attributes,CQA) 建立系统的风险质控体系,是对产品属性、生产工艺和产品质量之间关系的深刻理解[10]。开展注射剂一致性评价即是按照QbD的理念,从参比制剂的选择、处方工艺、原辅包质量控制、产品质量研究与控制以及稳定性研究等方面建立风险评估体系,分析注射剂质量风险来源,最终达到与参比制剂质量一致的要求。
3.1关于参比制剂的选择
参比制剂的正确与否直接影响到产品技术审评,属重大风险项。申请人应全面了解已上市注射剂的国内外上市背景、安全性和有效性数据、上市后不良反应监测情况,评价和确认其临床价值。
3.2处方和工艺技术要点
3.2.1处方
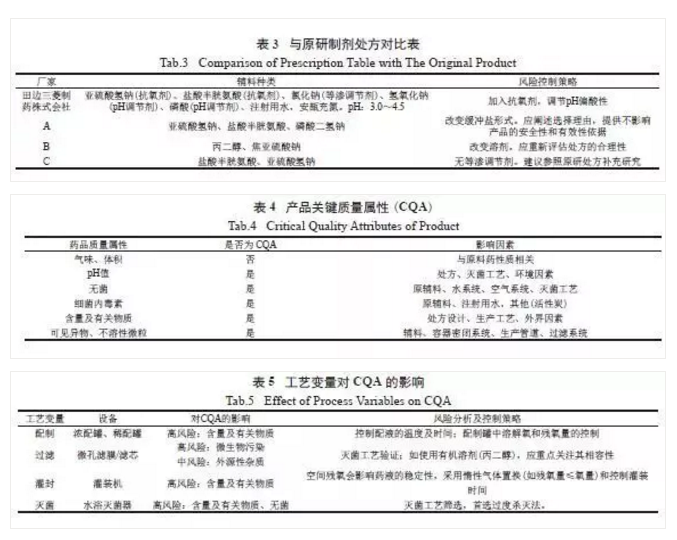
根据《技术要求》的要求,注射剂仿制药中的辅料种类和用量通常应与参比制剂相同,可接受范围为95%~ 105%。如附带专用溶剂,应与参比制剂的专用溶剂处方一致。一般而言,注射剂辅料的种类和用量情况可直接从药品说明书中获取,因此处方筛选的重点应该是通过建立合适的评价指标,结合试验对原研处方合理性分析。建议参考FDA指导原则《ANDA Submissions-Refuse-to-Receive Standards》。处方变更可明显影响产品质量,也是审评关注的重点,建议与原研处方保持一致。
3.2.2 工艺研究
建议采用与原研制剂尽可能一致的生产工艺,主要参考文献为:原研制剂专利、已公开的文献资料、部分产品说明书( 如灭菌与除菌技术等) 等,合理分析原研药处方组成( 抑菌剂、稳定剂等) 并全面剖析原研药品的工艺。在质量一致性的前提下,采用比原研制剂更加合理的工艺。
3.2.3 工艺验证及工艺控制
工艺验证:①对于终端灭菌药品,至少进行并提交灭菌或无菌工艺验证。②需提供工艺验证资料,包括工艺验证方案和验证报告。研究方案应结合处方进行考虑,在针对特定品种时还应考虑配液pH值、助溶剂等对直接接触的滤芯、硅胶管的影响。《技术要求》明确要求了包装的密封性验证、保持时间的验证,进一步加强药品生产工艺与质量保证的要求;同时要求除菌工艺生产的过滤前药液微生物进行监控,而且有需要定入中间体质量控制的趋势,一旦超过微生物负荷验证的范围,其产品的放行可能需要进一步的风险评估。③工艺控制中,建议明确关键工艺参数及其制定依据。④建议注册批与商业化批为同一生产线生产,批量应符合《技术要求》。
3.3 原辅包质量控制
对于注射剂仿制药而言,通常原料药可能会有多个来源,建议对不同来源的原料药进行质量对比研究,优选质量较好的,且应提供供应商审计情况。如原料药合法来源及质量控制的详细资料,或者原料药DMF 编号和资料以及原料供应商同意使用DMF 的授权书。此外还需关注原料药是否符合注射剂的要求:如采用自行精制的注射级原料药投料,就需给出精制工艺选择依据、详细的精制工艺及其验证资料、精制前后的质量对比研究资料等,以判断精制产品的质量控制是否符合注射用要求[13]。
3.4 质量研究与控制的技术要点
《技术要求》指出应根据QTPP 确立剂的CQA,建议完善的质量标准。同时应重点关注以下4 类杂质研究。
3.4.1 有关物质
应重点关注降解杂质,包括可能与处方中辅料如抗氧剂或包材形成新的杂质。建议研发者应对国内外药典收载的杂质进行全面分析,对所采用的原料药从合成工艺路线进行详细的分析,区别工艺杂质、降解杂质,原料药的杂质谱应小于或等于原研制剂杂质谱,对于新增杂质,应根据ICH Q3A 和3B 进行合理分析,区别报告限、鉴定限和质控限。
3.4.2 异构体杂质
对于存在几何异构体和手性异构体等情况,根据产品特点和生产工艺等方面的研究,确定是否订入标准。如活性化合物为手性化合物,应在制剂稳定性研究中开展对映异构体杂质的研究,积累批数据,酌情考虑是否订入质量标准。
3.4.3 遗传毒性杂质
建议参照ICH M7 对制剂产品可能存在的基因毒性杂质进行评估。
3.4.4 元素杂质
根据ICH Q3D 的规定,通过科学和基于风险的评估来确定制剂中元素杂质的控制策略,包括原辅包、生产设备等可能引入的元素杂质,特别关注生产管道( 如锰、镍等金属离子)、硅胶管等与药液的相容性。但也有研究认为,硅胶管作为药液过滤、灌装管道,与药液短暂接触,相容性试验中的相互作用可忽略[16]。以依达拉奉注射液为例,对原研日本IF 文件中收载的杂质分析见表5[11]。

3.5稳定性研究技术要点
配伍试验中,至少1 批为近效期样品。最好参照ICH Q1B 要求进行光照稳定性研究。光照试验的总照度不低于1.2×106 Lux·h、近紫外能量不低于200 W·h·m-2。建议研究者稳定性研究条件参考2015 年2 月CDE 颁布的《化学药物( 原料药和制剂) 稳定性研究技术指导原则》和ChP 2015 来制定。自制样品的贮藏条件应不低于原研制剂。
四.特殊注射剂一致性评价的关注点
目前上市特殊注射剂主要有脂质体、乳剂、混悬剂、微球及纳米粒等,由于其特殊的功能性辅料( 磷脂、PLGA 等)、多相体内外释放曲线、不同工艺过程对其释药行为均会产生影响,同时,也缺乏药典收载的体外评价方法、体内外相关性评价方法以及对于释药机制完全的了解,因此,特殊注射剂的一致性评价常面临极大的挑战。例如对于脂肪乳注射液的仿制,应尽量选择与原研制剂相同的卵磷脂辅料,优选同一种类和型号的卵磷脂辅料。对于载药脂肪乳,如果选择与原研制剂不同的卵磷脂,应针对来源、纯度和组成的不同进行比较试验,论证可能存在的对药品质量及安全有效性的风险[17]。
五.展望
注射剂由于其给药特点,各国药监部门均将其视为风险程度最高的品种之一。开展注射剂仿制药一致性评价,将会建立和健全注射剂仿制研发的风险质控体系,全面提升我国注射剂仿制的质量水平。对于特殊注射剂而言,由于其技术难度较高、生产工艺复杂,可借助特殊注射剂的一致性评价工作提升企业的制剂平台能力建设,增强企业的竞争力。
作者简介:
刘晓丹,博士,从事药品技术审评和核查工作。
陈桂良,博士生导师,从事药品审评核查工作。
文章来源:制药联盟者
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。