扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 新修订《药品注册管理办法》坚决落实党中央、国务院改革部署和新制修订法律精神。巩固42号文件和44号文件部署的改革成果,对药品注册管理进行调整完善,优化审评审批工作流程,建立科学高效的审评审批体系。
新修订《药品注册管理办法》坚决落实党中央、国务院改革部署和新制修订法律精神。巩固42号文件和44号文件部署的改革成果,对药品注册管理进行调整完善,优化审评审批工作流程,建立科学高效的审评审批体系。
此次修订突出药品注册的管理属性,围绕明确药品注册管理工作的基本要求,对药品注册的基本制度、基本原则、基本程序和各方主要责任义务等作出规定,未对科学技术要求作出规定。相较于2007年的《药品注册管理办法》附件,可谓是对技术要求的一种“解放”,有利于随着技术发展不断调整完善。
此次修订工作对既往国内药品注册管理的理念、思路以及程序设计等也进行了调整,将改革成果融入其中,增加了一系列新制度、新理念。
具体包括:药品上市许可持有人制度、药物临床试验默示许可制度、优先审评审批制度、原辅包关联审评审批制度、沟通交流制度以及专家咨询制度等,在程序上也提出药品注册检验前置、药品注册现场核查和上市前药品生产质量管理规范检查衔接实施等新理念。
新旧《药品注册管理办法》对比研究
2007年版《药品注册管理办法》共十四章,一百七十七条,六个附件。新修订《药品注册管理办法》共十章,一百二十六条,内容上进行了大幅缩减,原《药品注册管理办法》附件均不再保留。
除上述新制度新理念外,着重对比新旧《药品注册管理办法》在事权、药品注册分类、药物临床试验管理、药品上市许可路径、变更管理及工作时限几方面内容。
事权调整
原《药品注册管理办法》中规定:
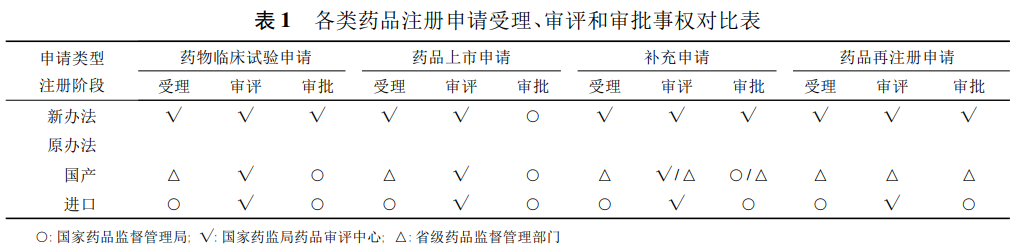
对于国产药品的新药临床试验、新药生产、仿制药申请均由省级药品监督管理部门受理,国家药品监督管理局(NMPA,以下简称国家局)药品审评中心进行审评,最终由国家局审批;
对于进口药品申请,由国家局受理,国家局药品审评中心审评,最终由国家局审批;
对于补充申请,在原办法附件4中详细分为36项补充申请事项,并分别报国家局审批、省级药品监督管理部门审批后国家局备案、国家局备案或者省级药品监督管理部门备案;
对于药品再注册申请,国产药品由省级药品监督管理部门审批,进口药品由国家局药品审评中心审评,国家局审批。
2017年4月,原国家食品药品监督管理总局发布《关于调整部分药品行政审批事项审批程序的决定》(总局令第31号),其中规定自2017年5月1日起将由总局审批的药物临床试验审批决定(含国产和进口)、药品补充申请审批决定(含国产和进口)以及进口再注册审批决定调整为由总局药品审评中心以总局名义作出。
同年11月发布《关于调整药品注册受理工作的公告》(2017年第134号),其中规定自2017年12月1日起将省级药品监督管理部门受理,总局审评审批的药品注册申请调整为总局集中受理。
新修订《药品注册管理办法》延续了药品审评审批改革期间对受理审评审批程序作出的调整和优化。
对于药物临床试验申请、上市许可申请、补充申请及进口药品再注册申请,均由国家局药品审评中心受理,除上市许可申请由国家局审批外,药物临床试验申请、补充申请和进口药品再注册申请均由国家局药品审评中心以国家局名义作出审批决定。
国产药品再注册申请仍由省级药品监督管理部门受理和审批。与2007年的《药品注册管理办法》相比,各类申请受理、审评和审批事权的调整详见表1。
药品注册分类的调整
原《药品注册管理办法》附件中分别对中药和天然药物、化学药品和生物制品的注册分类作出规定,其中中药和天然药物分为1~9类,化学药品分为1~6类,治疗用生物制品分为1~15类,预防用生物制品分为1~15类。
2016年原国家食品药品监督管理总局发布《关于发布化学药品注册分类改革工作方案的公告》(2016年第51号)将化学药品的分类调整为5个类别。
新《药品注册管理办法》中规定:中药注册按照中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等进行分类;
化学药注册按照化学药创新药、化学药改良型新药、仿制药等进行分类;
生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药)等进行分类,细化药品注册分类根据注册药品的产品特性、创新程度和审评管理需要组织制定并向社会公布。
新分类方式是按照药品的创新程度进行划分,与国家鼓励创新的各项改革制度契合,在一定程度上能够促进药物研制创新。
上述分类中也蕴含了简化审评审批路径的分类,如中药的古代经典名方中药复方制剂、化学药的仿制药以及生物制品中的已上市生物制品(含生物类似药)。
此外,此次新《药品注册管理办法》不再区分进口药与国产药,而是使用境外生产和境内生产,以不同的场地进行区分,这有利于统一进口与国产药品的标准和程序。
药物临床试验管理的调整
新《药品注册管理办法》规定的临床试验范围是以药品上市注册为目的,为确定药物安全性与有效性在人体开展的药物研究。
在临床试验分期上,按照药物研发阶段分为Ⅰ期~Ⅳ期和生物等效性试验;按照研究目的分为临床药理学研究、探索性临床试验、确证性临床试验和上市后研究。
对于不同分期以及不同研究内容的定义,则参照ICH相关指南中的具体定义和描述,对于临床试验开展过程中的具体技术内容不再正文中做要求,而是对临床试验审评审批的流程以及临床试验获准后的管理作出了进一步要求。
如对于药物临床试验申请实施默示许可、生物等效性试验实施备案等审评审批流程,临床试验期间的各类报告、临床试验期间变更的申报流程以及临床试验的暂停、恢复及终止程序等。同时还增加了临床试验登记的相关要求。
上述临床试验管理的程序和要求都是与原《药品注册管理办法》有所不同的。
药品上市许可程序的调整
新《药品注册管理办法》在药品上市许可程序上设定了3种路径:完整路径、直接上市路径和非处方药上市路径,不再按新药、仿制药和进口药划分上市路径。
其中,完整路径是指在完成支持药品上市注册的药学、药理毒理学和临床试验等研究后,提出上市许可申请;
直接上市路径是指经申请人评估无需或者不能开展药物临床试验,符合豁免药物临床试验条件的,申请人可以直接提出药品上市许可申请,如仿制药、按照药品管理的体外诊断试剂等;
非处方药上市路径是符合新《药品注册管理办法》第三十六条规定情形的非处方药,可以直接提出上市许可申请。
药品变更管理的调整
原《药品注册管理办法》中的变更是按照是否影响药品质量进行划分,并在附件中规定了36种变更情形。新《药品注册管理办法》在变更管理上首先按阶段进行划分,即临床试验期间的变更、审评期间的变更以及上市后变更。
临床试验阶段的变更主要是按照对受试者安全影响的程度不同,对应不同的管理路径,其中影响受试者安全的变更,以补充申请的方式提交变更申请,实施60d默示许可;不影响受试者安全的变更,在研发期间安全性更新报告中报告。
审评期间的变更主要是按照对原注册申请可评价性的影响,如发生可能影响药品安全性、有效性和质量可控性的重大变更,则可能导致原注册申请不具备可评价性,需要申请人撤回原注册申请,补充完善后重新申报。
上市后变更主要是按照对所生产药品的安全性、有效性和质量可控性的影响程度,实施分类管理,分为审批类、备案类和年度报告类,对应生产过程中的重大变更、中等变更和微小变更。
从对药品变更的管理上讲,更加科学和细化,也体现了国务院“放管服”的要求。
工作时限的调整
1)审评时限的调整
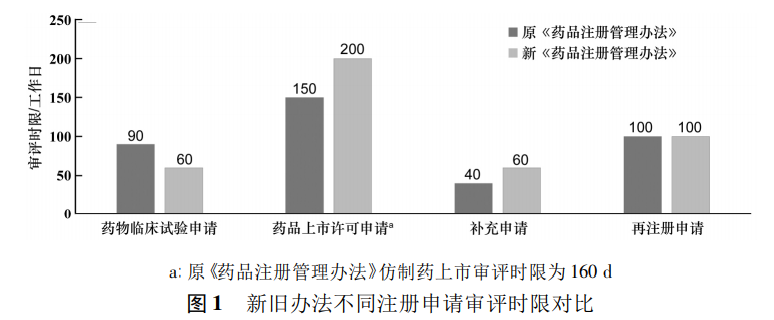
新《药品注册管理办法》在药品注册审评时限上作出相应调整,其中药物临床试验的审评时限由原来的90d缩短至60d,药品上市许可申请和补充申请有所延长,药品再注册的时限没有发生变化,详见图1。
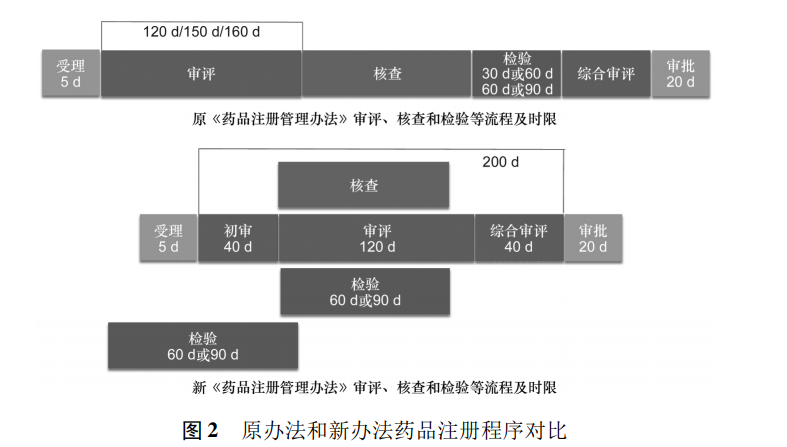
4)其他时限的调整
新《药品注册管理办法》增加了药品通用名称核准和非处方药适宜性审查的时限要求,分别为30d,进一步明确了药品注册各环节所需的时限要求。
综上所述,新《药品注册管理办法》细化了药品注册各环节的时限,对于药品注册核查与检验还明确了启动和完成的时间节点,这些时限上的具体要求更加有利于行政相对人实现其时限的可预期性。
结语
落实新法精神,固化改革成果,鼓励药物创新
2020年新修订的《药品注册管理办法》落实了《药品管理法》《疫苗管理法》等法律的精神以及药品审评审批制度改革精神,将药品上市许可持有人制度、临床试验默示许可制度、优先审评审批制度以及附条件批准制度等固化。
继续实施鼓励创新的政策,设置了突破性疗法、优先审评审批、附条件批准和特别审批程序4个通道,加快药品审评审批,明确了4个通道的适用范围、纳入标准、支持政策和终止程序及相关要求。
重设规章架构,拓展调整空间
2020年新修订的《药品注册管理办法》调整了框架结构,与2007年的《药品注册管理办法》相比,此次修订的办法中不再设置附件,对中药、化药、生物制品的注册分类和申报资料要求,以及对补充申请和药品再注册申请的具体要求,均以配套文件的形式发布,主体文件仅对药品注册的基本制度、基本程序、基本要求等作出规定,这种结构设置有利于随着科学技术的发展调整相应的申报资料要求。
遵循规律,优化程序,提高效率
2020年新修订的《药品注册管理办法》优化和调整了药品注册程序,尤其是审评、检查检验等各环节的衔接上,比如增加了直接申请上市的途径、药品注册检验可以前置到受理前,药品注册检查可与GMP检查同步开展,增加了检查检验启动时间和衔接要求,增加了补充非技术资料的途径等。
这些程序上的优化,在一定程度上能够提升药品审评审批工作效率。同时,在程序设置上也更加遵循药物研发规律,比如在药品注册申请前或临床试验开展的过程中,可以与技术审评部门进行沟通交流,对临床试验期间的变更和上市后的变更实行分类管理,分别根据对受试者安全性影响程度和对药品生产过程的影响程度实行审批、备案或者报告。
加大信息公开,落实社会共治
2020年新修订的《药品注册管理办法》增加信息公开的力度,提高了药品审评审批工作的透明度。增加了对临床试验结果信息的公开、对批准上市药物审评结果和依据的公开、对说明书的公开以及对监督检查发现的违法违规行为的公开并及时更新等。
药品审评审批制度改革仍在路上,在新《药品注册管理办法》落地实施的过程中可能还会出现各种新问题新情况,需要监管部门和行政相关人员在工作实践中加强沟通,共同研究,进一步夯实改革成果,推动药品注册工作迈上新台阶。
文章来源:《中国新药杂志》
原标题:新版《药品注册管理办法》修订内容研究与思考
作者:王婧璨,张晓东,温宝书,蒲嘉琪(国家药品监督管理局药品审评中心,北京)
文章来源:制药联盟者
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。