扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 QC 实验室“数据完整性”
QC 实验室“数据完整性”
检验记录数据完整是药品检测体系中最基本的要求,数据完整性( Data integrity) 体现在数据的一致性、真实性和完整性三个方面。当前我国医药生产企业飞行检查中QC 实验室数据完整性方面出现问题较多,回顾药品检验记录“数据完整性”的来龙去脉,同时理解当前FDA 对“数据完整性”的本质要求,对解决如何更好的完善QC 实验室数据完整性具有很好的指导意义。
1.数据完整性的概述:
。
1.1数据完整性定义
药品生产质量管理规范(2010年修订)附录11:是指数据的准确性和可靠性,用于描述存储的所有数据值均处于客观真实的状态。
MHRA 数据完整性指南定义:数据完整性的范畴包括所有数据在整个数据生命周期中的全面性、一致性和准确性的程度。
CFDA:数据管理应贯穿整个数据生命周期,坚持真实、准确、及时、可追溯的 数据管理原则,确保数据可靠性(Data Integrity)。
1.2数据完整性要求:ALCOA
A:attributable to the person generating the data 可追溯至数据由谁生成
L:legible and permanent 清晰并持久
C:contemporaneous 同步
O:original (or true copy) 初始(或正确的副本)
A:accurate 准确
2. 数据完整性的法规要求:
FDA: Data Integrity and Compliance With Dmg CGMP Questions and Answers Guidance for Industry 数据可靠性与药品CGMP合规问答
MHRA:GMP Data Integrity Definitions and Guidance for Industry
GMP数据可靠性定义和工业指南 GXP Data Integrity Guidance and Definitions GXP数据可靠性指南和定义
PDA:Elements of a Code of Conduct for Data Integrity in the Pharmaceutical Industry 制药行业数据完整行为准则要素
PIC/S: Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments GMP/GDP监管环境下数据管理和可靠性的良好实践
NMPA:药品数据管理规范
WHO 数据完整性指南要求
EMA 数据完整性问答
CFDI 药品数据管理规范
PDA 实验室数据完整性管理
3.QC 实验室数据完整性常见缺陷:
QC 实验室作为药品质量的把关部门,检验数据真实、完整与否关系到药品的安全。QC 实验室是数据完整性飞行检查中关注的重点部门。QC 实验室数据完整性缺陷集中于两方面〔9〕: 一是来自于人工观察、填写的纸质记录; 二是来自于仪器、设备通过复杂的计算机化系统产生的图谱或电子记录。
3. 1 人工观察填写的纸质记录
实验室纸质记录主要包括:①取样记录; ②检验记录或实验室工作记录本、报告; ③实验室日志、检验台账、仪器维护和使用日志、色谱柱使用记录、标准品使用记录。这些记录是QC 实验室“数据完整性”的基本支撑材料。检验人员保留有原始支持性数据,证明原料药符合既定预期的质量标准,是质量部门的基本职责。飞行检查过程中纸质记录的数据完整性缺陷常见如下情形:
3.1.1 未对文件和记录版本( 变更) 进行控制
实验室缺乏对文件和记录的有效控制,没有保留全部关键的原始数据。如检查发现某制药企业QC 实验室发现OOS( Out Of Specification)后,进行重新取样,重新进针和重新处理等操作。公司没有对OOS 的结果进行调查,也没有记录解释为什么对不符合要求的中间体,成品等进行重新取样和重新测试,最后没有报告OOS 结果,只报告了重新测试得到的合格结果。在对QC 微生物实验室的检查中,发现批号为的片剂申报批次样品微生物检验于年6 月13 日进行。企业未能向检查员提供记录培养时长和所用培养基的原始数据,检验员说该批次整个微生物检验已经在上周完成了,但其“忘记”在记录本上记下这些详细信息。
3.1.2 未对原始空白记录和空白记录的发放进行控制
对原始记录的发放和空白记录方法不做控制,检验人员可以轻松地拿到新的原始空白记录和不受约束的处理原始数据记录表格。如检查中发现QC 实验室不受约束的原始数据记录表来记录原始数据。
3.1.3 未对已填写记录的变更进行控制
公司无法确保包括所有检测数据在内的实验室记录证明产品符合质量标准,毁坏或缺少原始的实验室数据。如检查中发现原始数据记录存在缺页,检查员询问得到工作人员答复是检验记录被误撕了。
3. 2 仪器、设备通过复杂的计算机化系统产生的图谱或电子记录
3.2.1 未能用电子数据处理系统或其他可靠方式记录的数据资料
检查员在控制GC 的电脑发现原始数据被删除,GC操作软件允许检验员从电脑中删除原始数据,且没有审计追踪信息或其他形式的数据追踪。该软件对同一文件夹内原始数据文件采用流水号的方式进行命名,当一个原始文件被删除或移除出文件夹后,其后面的文件就以该删除文件的序号进行命名。即使这些文件不是按操作顺序生成也无法看出来,因此可以对数据进行操作。
此外,检查员发现QC 实验室某样品色谱图经过手动积分处理,检验员也承认对色谱图积分数据进行修改,但该实验室没有规范的标准操作程序。
3.2.2 未保存检验设备和仪器待确认和校准记录:
检查中检查员发现编号为F1-002 的天平校验记录被撕毁。检验员解释说由于采用了错误的重量进行校验,校验人员随后重新进行了校验并丢弃了之前的校验记录。此外,检查员还发现另一台天平的原始校验记录也被丢弃了。
3.2.3 未能对计算机或相关系统权限进行控制:
QC 人员在计算机系统上未经监管及创建了未经授权的文件夹。如检查员对QC 实验室HPLC 的EMPOWER Ⅲ数据管理工作站里2013 ~ 2014 年数据进行了审核,发现了一个名为“WASH”的文件夹,根据实验室的管理,该文件夹是用来保存进样前后空白溶剂进行柱清洗的信息,但实验室没有SOP( Standard Operation Procedure) 详细说明该程序。一名检验人员声称该文件夹不包含任何标准品或样品的进样结果,然而检查员发现该文件夹共有3353 针结果,有些显示为样品进样结果。
检验员随后承认在“WASH”文件夹中有一个文件名为“19”的单独进样结果。实验室也承认检验员未能遵守“实验室工作通则”所要求的良好实验室行为规范,且该检验员做出的进样决定未经授权和批准。企业声称在事件发生6 个月以后对该检验员进行了面谈,认为可能是由于其无意间使用了液相托盘中的一个旧样品瓶用于柱冲洗,检查员质疑实验室仅基于6 个月前可能发生事件的记忆得出该结论,而没有任何支持性文件或记录。
4.QC 实验室“数据完整性”的保障思考:
药品质量优劣的关键在于生产,而非在于检验。基于“诚信”生产药品为基础,做好QC 实验室“数据完整性”应注重结合以下四点:
4. 1 完善数据与记录管理的技术基础,完善记录管理程序
数据与数据管理基础在于建立完善的数据库系统,对药品检验过程中每一个环节统一编码,同时同步推进代码管理系统维护,保障检验原始数据可追溯性的实现〔8〕。如对实验室物料统一编制物料代码,确保代码与标示系统记录物料一致。如取样单、标签、检验状态信息卡等现场标示信息应一一对应。建立完善的文件、检验记录的编号与控制规范,建立QC实验室台账、检验日志管理规范,严格落实实验室检验空白记录和空白记录的发放处于受控状态,实现检验数据追溯性和完整性。
实验室应明确检验记录管理的范围控制,包括记录管理流程从而实现完整性、准确性、真实性、及时性与一致性。对检验记录管理应指定人员负责,定期检查,同时规定记录的填写要求,记录更改要求,记录的复核要求和记录的保存要求( 地点、期限) 。形成并实现检验记录及时、完整归档,形成完整,一致,长久,可获取的原始检验记录管理机制。
4. 2 完善记录文件
记录是程序执行的载体,人用药物注册技术要求国际协调会(ICH) Q7A 第6. 6 条对实验室控制记录做了要求。记录设计应包括生产质量控制管理活动执行的信息如质量控制活动的时间、地点、执行人、执行过程、执行结果等信息; 对关键执行过程的信息要双人复核,关键执行过程的审核、批准、执行后确认等信息应一一记录在案。同步执行后的回顾与评估,有任何异常现象与结果应一一记录。
4. 3 建立计算机化系统管理系统
电子记录是指任何文本、图表、数据、声音、图示的或其他的以电子形式表现的信息的混合,它的建立、修改、维护、归档、检索或分发是由计算机系统来完成的。电子签名是指一种由一个人执行、采用或批准成为与其个人的手写签名具有相同的法律效力的计算机数据的任意符号或一系列符号的编译。电子签名和电子记录的本质是一样的,都是电子形式的计算机数据组成的。建立计算机化管理系统,启用审计和追踪功能,对原始数据保存采用动态化与静态化相结合的数据储存方式,实现数据可追溯性。
4. 4 良好的员工GMP意识与行为
检验原始记录应正确及时让检查人员获取,是谁完成检验,做了哪些检验项目以及完整的原始的检验数据。检验记录严禁使用涂改液,检验记录严禁出现记录不完整或不准确现象,严禁提前填写记录,严禁操作未完成前填写记录或签名,避免操作人未签名的情况下,复核人已签名。
药品质量检测体系中最基本的要求是检验数据记录真实完整,真实的检验记录是判断药品质量安全与否的关键。QC检验员作为药品质量的把关人,出具的实验结果要经得住检验,其检验流程也要经得住考验,这些都需要通过实验室检验原始记录来呈现。真实完整的检验记录既能反映药品的质量,也能反映制药企业的管理水平。做好检验原始数据完整性是QC 实验室的愿望、是生产企业的愿望、也是用药患者的愿望。
微生物实验室数据完整性的关注重点:
工作流程
数据完整性关注点
取样
内毒素取样采用不恰当的容器
取样前的消毒
在线取样工具灭菌和冷却
传递窗的紫外
样品至于冰箱保存
A级区消毒剂的验证,消毒对样品的影响
样品转移
丢失、污染、混淆
样品处理
跌落、损坏、污染、混淆
样品培养
温度超限或超时
读取培养
结果
发生漏、误、提前记录
超标数据
报告
未及时报告
数据计算
稀释度错
配制记录、
设备日志记录
测试计划和平皿数据一致性,培养基配制数量、培养箱数量是否够
声称进行微生物测试,但缺乏相应的设备(培养箱)和所需的培养基 使用记录
未能记录和报告准确的环境监测数据情况,检查时发现有沉降菌情况
,平时记录都是无
产品无菌测试失败没有调查和记录,使用非正式记录本来记录微生物 污染情况,但没有相应调查
原始记录
是否存在某检验员短时间内测试量/记录(worksheet) 过多情况。
核对检验员/复核人笔记、签名;时间和考勤匹配等情况。
质量控制日志:日志是数据可靠性问题的重灾区,FDA在日志中的
发现五花八门,包括:日志填写不及时、不规范,日志修改、管理 不受控,日志暴露企业调查不充分等问题培养基模拟灌装记录不真 实,包括培养基促生长试验微生物没有生长也通过试验。检查员开 始关注培养基因空气采样而留下的痕迹。
检验记录
检验记录相关记录的复核,如设备使用记录
实验室某个实验员出具的检验记录数量远远超过理论上一个单独 实验员一天或一周正常的数量;
积压的检验记录作为批次记录在月底被审核和批准;
检测记录上签字的人员在检测当天并未在岗;
检验记录被未授权的审核/批准人员审核或批准;
书写或签名处有涂改;
报告形式变更,例如用0替代<1cfu;
报告的结果几乎接近标准值;
检验记录填写过于完美,可能为重新誊抄的记录;
在培养基适用性检查之前就使用该培养基(未关注最终结果);
记录表格丢失或编号不连续;
记录表和调查报告之间不一致;
同一批次报告2次结论。
合同实验室
资质确认
进行现场审计
以及提供给合同实验室一些样品进行测试判断其测试水平和数据完整性
合同实验室的数据完整性
OOS的报告
设备和
仪器检查
仪器的清洁和维护
设备的校准
鉴定仪器中鉴定数据的追溯
设备故障的记录与使用情况
环境监测
空气采样、采样头、沉降碟、最终计数不科学等;
可能出现的数据造假:
实验室某个实验员出具的检验记录数量远远超过理论上一个单独实验员一天或一周正常的数量;
月底开始集中审核和批准积压的一批次样品检验记录;
检测记录上签字的人员在检测当天并未在岗;
未授权的记录审核员;
书写或签名处有涂改(记录可以修改,但是记录修改应符合相关文件规定,一般规定在原纪录处划线,原纪录上方填写正确记录,然后签字并注明日期,必要时还应注明修改原因);
报告形式变更,例如用0替代<1cfu;
报告的结果几乎接近标准值;
OOS结果报告周期的变更;
刚好填满一整张记录,可能显示为重新誊抄的记录;
在培养基适用性检查之前就使用该培养基;
记录表格丢失或表格编号不连续;
记录表和调查报告之间存在差异;
同一批次报告2次结论。
平皿计数结果发生错误 , 培养结果与纸质记录不一致 ,重复进行微生物鉴定以取得良好的数据,菌落特征与鉴定结果相冲突 – 培养条件超范围后未对培养结果进行评估
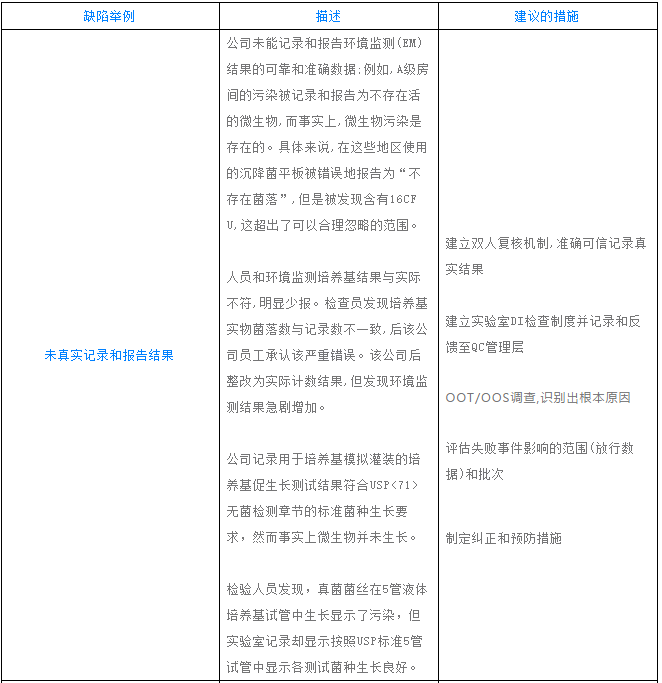
数据完整性缺陷举例和分析



文章来源: CROU制药在线
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。