扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们
 细胞疗法根据所治疗的适应症、细胞类型以及细胞是取自和施用于同一个体(自体)还是源自健康供体(同种异体)进行分类。细胞疗法的监管分类区分了用于同源使用、移植或输血的最低操作的细胞限度,以及作为药物进行采集操作和管理的细胞。临床医学中细胞疗法必须符合质量、安全性和有效性标准才能获得上市许可。此类疗法可细分为体细胞疗法、基因疗法和组织工程产品。它们可以由自体或同种异体来源制造,并含有非细胞成分(例如,化学或生物化合物和基质)。
细胞疗法根据所治疗的适应症、细胞类型以及细胞是取自和施用于同一个体(自体)还是源自健康供体(同种异体)进行分类。细胞疗法的监管分类区分了用于同源使用、移植或输血的最低操作的细胞限度,以及作为药物进行采集操作和管理的细胞。临床医学中细胞疗法必须符合质量、安全性和有效性标准才能获得上市许可。此类疗法可细分为体细胞疗法、基因疗法和组织工程产品。它们可以由自体或同种异体来源制造,并含有非细胞成分(例如,化学或生物化合物和基质)。
现成的同种异体产品基于从健康供体获得的细胞,并通过基因编辑进行修饰以消除免疫排斥反应等功能。修饰的细胞被扩增以产生大量的产品,这些产品可以冷冻和储存,直到患者需要为止。患者特异性细胞疗法通常是自体的,但也可以是来自匹配供体的同种异体细胞疗法,以防止免疫排斥。患者特异性细胞疗法和现成产品之间的主要区别在于,前者需要为每位患者提供独特的制造批次。基因编辑已被证明可成功用于商业化患者特异性细胞疗法,例如使用嵌合抗原受体T(CAR-T)细胞进行过继细胞转移(如下图1)。
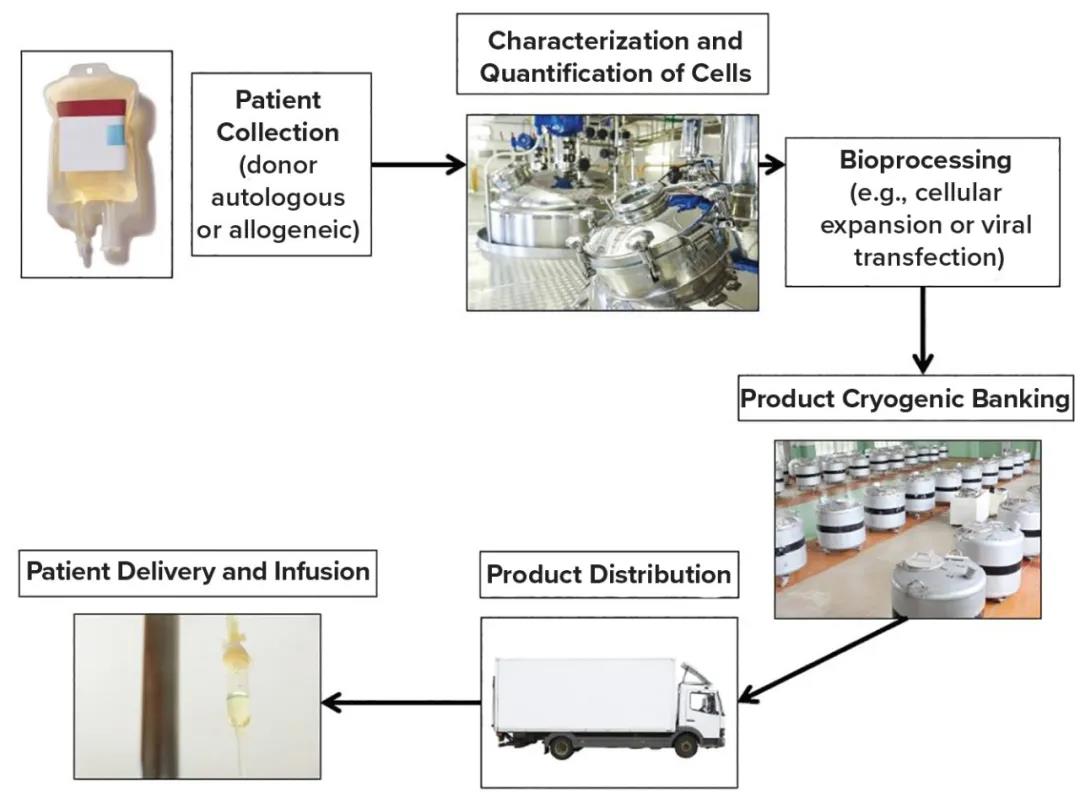
图1:细胞疗法的生产使用流程图
监管要求
最低限度采集细胞的操作治疗产品的监管已经颁布。这些法规侧重于预防传染病的传入和传播。要求在企业注册、捐赠者资格标准和处理细胞产品都必须遵守CGTP(Current Good Tissue Practice)。这些产品作为药品、医疗器械和生物制品受到监管,这增加了CGMP(Current Good Manufacturing Practice)条件下制造这些产品的监管要求。随着细胞疗法从临床转向商业制造,监管机构使用基于风险的方法制定法规。正在制定允许生产安全有效的细胞治疗产品的法规。仍然需要科学和技术发展来帮助细胞疗法制造商应用CGMP条件制造和进行质量控制(QC)流程。
细胞来源
细胞的来源特性必须解决,以确保产品的一致性。应该由患者专用产品的制造企业提供详细的培训和专门的收集文件来解决这种可变性。由于来源细胞材料具有相当高的价值,因此收集或生产大量用于工艺开发和验证的细胞材料是一项行业挑战。有限的保质期和细胞数量会使QC测试和稳定性测定变得复杂。
必须对需要储存的细胞进行广泛的表征,以防止微生物或病毒制剂的潜在污染以及致瘤性。确定产品的关键质量属性(Critical Quality Attributes,CQA)并开发检测方法以监测其效力对于细胞治疗产品的商业化至关重要。源细胞和最终细胞产物的表征对于自体和同种异体疗法很重要。源细胞的特征基于表面标记的存在、细胞大小以及与细胞来源和作用方式相关的属性组合。最佳表征对于提高细胞特定群体选择的准确性至关重要。
自体或同种异体细胞疗法生产的GMP要求
自体和同种异体产品表现出不同的特征并提出不同的制造挑战。自体产品的挑战包括患者的变异性和对小规模制造批次的需求。由于所针对患者特异性,它们可以避免最后产品的免疫原性。同种异体产品就没那么复杂,因为它们通常使用具有安全特征的细胞库,并可以扩大规模生产用于大量接受者的通用产品。但此类产品的并发症包括潜在的免疫原性(来自受体,在这种情况下,细胞在给药后只能存活很短的时间,或在移植物抗宿主反应中)、潜在的肿瘤形成以及对适当的制造平台要求。
对于自体细胞疗法,患者既是原料来源又是接受者。因此,重点是收集、处理和重新分配来自个体患者和向个体患者提供的细胞。自体疗法有利于患者安全,因为它们消除了移植物抗宿主病的担忧。对于这些疗法,应使用封闭的制造系统来降低产品污染的风险。在收集细胞时应使用处理文件的个性化批次记录。材料应在整个产品生命周期内进行追踪,包括运输、制造和患者使用期间的管理。由于细胞疗法对时间敏感,因此医疗专业人员在收集地点使用自动化系统处理细胞是可行的。这个过程可能就包括了细胞收集。
对于同种异体细胞疗法,来自一个供体的细胞可作为治疗多位患者的源材料。如果细胞扩增的数量有限,细胞捐赠整个过程可能会很复杂,因此可能需要来自同一捐赠者的多次收集。同种异体产品制造对于自动化细胞处理更可行,因为一批中的所有细胞都来自通用供体。将生产从开发规模扩大到制造规模可能具有挑战性,因为它涉及细胞扩增、体积减小和收获的不同处理系统。用于扩大制造规模的制造设备,例如带有微载体的一次性生物反应器,无法产生与用于研究和开发的摇瓶或小反应器相同的产品效力。
必须仔细评估用于制造细胞疗法的原材料和组件的收集和加工。包括培养基、细胞附着基质、培养容器、管道、袋子和其他一次性用品。培养容器、管道和袋子等一次性组件需要测试可提取物和可浸出物,因为这些材料会危及细胞生长和活力以及患者安全。应使用重组血清蛋白代替动物或人源血清,以防止细菌和病毒污染的风险。
应完善供应商资格认证程序:通过供应商筛选、现场审核、质量协议和供应商监控。每一批新的组件材料都应进行测试,生物制造商应随附供应商生成的分析证书(Certificate of Analysis,CoA),以确保此类组件符合要求。
执行收集程序的医疗专业人员通常未受过CGMP法规培训。收集文件应成为CGMP记录的一部分,包括收集设备和现场测试生成的电子数据。所使用的器具制造地点取决于产品的类型和这些产品的应用。临床或集中制造模式之间的决定应基于产品适应症和产品稳定性。
从收集地点到制造地进行的细胞疗法生产需要在CGMP设施中进行严格控制。这包括所有制造空间、原材料和成品的存储仓库以及实验室区域。细胞治疗生产设施必须设计为无菌处理。开发全封闭制造设备和用于过程测试的内置控制是加工的最佳选择。必须使用质量源于设计(QbD)模型来考虑从临床到商业制造的工艺流程,以验证CGMP制造工艺。这涉及定义关键工艺参数(Critical Process Parameter,CPP)、工艺参数和过程中测试。此类信息应源自开发研究,并用于定义工艺验证的设计空间。作为持续过程验证的一部分,应持续监控这些参数和测试。
其他重要的验证和验证活动包括合格的制造设施和设备、开发设施和设备清洁验证、开发分析测试和方法验证、开发环境监测程序、开发批次记录以及制定持续的过程验证计划。这些验证和验证CGMP要求应通过使用可归因的、清晰的、同期的、原始的、准确的(ALCOA)文件(可溯源文件)、无菌工艺和监管纳入数据完整性(在制造和实验室测试中)来满足。
制造企业必须制定程序,以防止因原材料、环境条件和操作人员而引起的微生物和交叉产品污染。他们必须建立可靠的容器灭菌方法以及原材料和试剂的控制。使用高效微粒吸收过滤器和洁净室可以防止材料因环境污染物或其他产品而在空气中交叉污染。应监测温度、湿度和压差等参数,因为它们与颗粒生成和微生物增殖有关。
材料和人员流动应该分开并且是单向的,以尽量减少交叉污染。操作员应接受洁净室无菌处理技术的培训。
自动化处理是细胞疗法制造的最佳选择。灵活且适应性强的制造工艺可以最好地解决源细胞特性的可变性,以确保最终产品具有相同的高质量和一致性。可以基于CQA的实时测量来控制过程中的培养条件。CQA的在线检测和测量以及效力、功效和安全性参数的快速测量的集成是自动化细胞制造过程的基本要素,并将有助于建立完善的CQA。
每个产品批次都应通过特定于该产品特性的特定测试。应在适当的情况下进行致瘤性和生物相容性测试。广泛的产品表征对于过程验证和过程中和放行测试规范的开发至关重要。
细胞疗法制造不允许对最终产品进行最终灭菌或通过过滤去除微生物污染物。因此,测试起始材料并验证无菌制造工艺以确保产品不受污染非常重要。应尽可能在产品上市前进行无细菌、支原体和真菌的无菌测试。一些细胞治疗产品需要完成所有必需的药典测试之前就向患者给药。在这种情况下,可以使用快速测试方法。例如,基于聚合酶链反应(PCR)的支原体检测和快速微生物检测方法已用于细胞疗法的放行测试,因为这些方法能够及早测试出潜在的微生物污染。
活细胞是动态的。它们可以继续在人体内生长、分化、迁移和相互作用。给药前细胞产品的表征并不能完全描述细胞治疗后患者细胞的表型和基因型。特征和CQA应根据患者数据的反馈和对治疗后监测患者作用机制的更好理解重新定义。使用治疗后患者数据来了解最有效的治疗细胞可以减少治疗和制造时间表所需的细胞数量。
药物分发程序
大多数细胞疗法在环境温度下无法长时间保持活力。这适用于同种异体和自体细胞疗法。患者的细胞必须被运送到处理设施并返回进行患者治疗时,自体细胞疗法面临额外的挑战。
可以从单个中央制造基地供应足够稳定以进行全球运输的同种异体产品,从而提供生产的效率和一致性。然而,稳定性有限的产品(自体和同种异体)可能需要其他式模型,其中制造发生在多个生产中心,甚至在不同的处理地点。必须验证细胞治疗产品的运输并监测温度。监管链和细胞环境条件记录应从收集一直持续。
细胞疗法的潜力通过解决疾病的根本原因、改变病程和逆转已经发生的损害,细胞疗法的使用可以超越传统的疾病治疗。从发现到研究和开发到商业制造产品的转变需要生物制造企业将CGMP法规纳入这些产品的收集、生产和交付中。这种新型再生医学的CGMP法规将由临床、工业和监管机构共同制定。这样的发展将使细胞疗法越来越多地被患者接受,并将为许多疾病提供新的治疗方法(也许是治愈方法)。
文章参考:
1、42 US Code §262: Regulation of Biological Products, 2010. US Government Publishing Office: Washington, DC.
2、EudraLex: The Rules Governing Medicinal Products in the European Union Volume 4 — EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 2, Manufacture of Biological Active Substances and Medicinal Products for Human Use. European Medicines Agency: London, UK, 2012.
3、Guide to the Quality and Safety of Tissues and Cells for Human Application. European Directorate for the Quality of Medicines: Strasbourg, France, 2017.
4、21 CFR Part 1271: Human Cells, Tissues, and Cellular and Tissue-Based Products. Office of the Federal Register, US Government Publishing Office: Washington, DC.
5、21 CFR Part 600: Biological Products. Office of the Federal Register, US Government Publishing Office: Washington, DC.
6、21 CFR Part 200: General Drug. Office of the Federal Register, US Government Publishing Office: Washington, DC.
7、Regulation (EC) No 1394/2007: European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) No 726/2004. European Commission: Brussels, Belgium, 2007.
8、Guidance for Industry: Human Somatic Cell Therapy and Gene Therapy. US Food and Drug Administration: Rockville, MD, March 1998.
9、21 US Code Chapter 9: Federal Food, Drug, and Cosmetic Act, Section 351. US Government Publishing Office: Washington, DC.
10、Common Standards for Cellular Therapies. Foundation for the Accreditation of Cellular Therapy: Omaha, Nebraska, March 2015.
11、Directive 2004/23/EC of the European Parliament and of the Council of 31 March 2004 (Human Tissue and Cells Standards). European Commission: Brussels, Belgium, 2004.
12、WHO Technical Report Series No. 986: Good Manufacturing Practices for Pharmaceutical Products — Main Principles, Annex 2. World Health Organization: Geneva, Switzerland, 2014.
13、ICH Q8(R2): Pharmaceutical Development. International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use: Geneva, Switzerland, August 2009.
14、Guidance for Industry: PAT — A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. US Food and Drug Association: Rockville, MD, 2004.
15、European Pharmacopoeia (PhEur), 9th ed. European Directorate for the Quality of Medicines: Strasbourg, France 2018.
16 USP 36–NF 31. US Pharmacopoeial Convention: North Bethesda, MD, 2013.
文章来源:抗体圈
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。