扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们

(接上)
4.1.5. Sterile filtration
除菌过滤
The filter data to be provided in the quality dossier is summarized in Table 3.
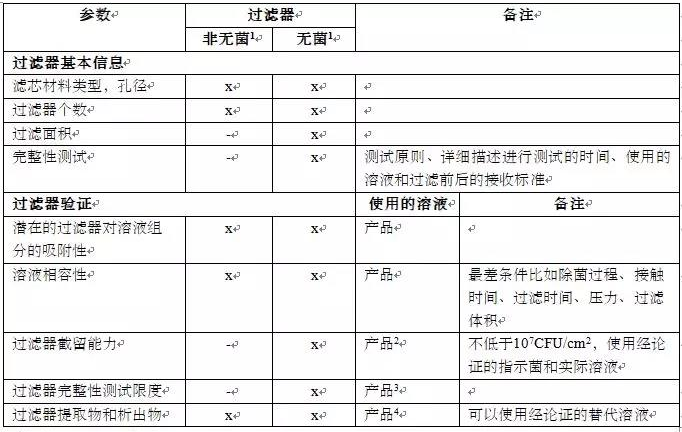
在质量文件中应提供的过滤器的数据总结在表格3中。
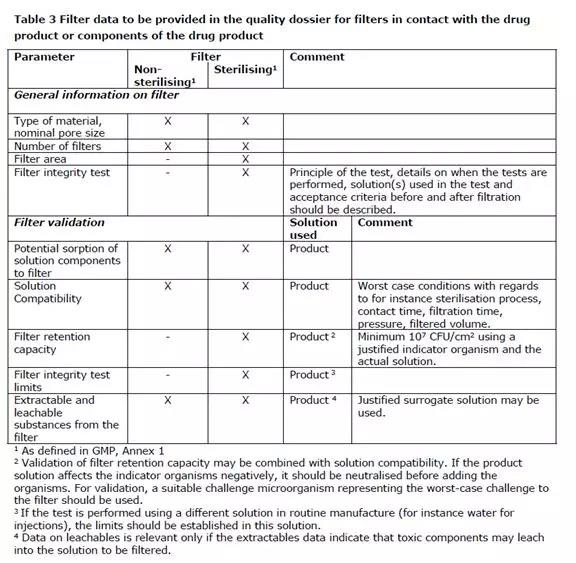
The integrity of the sterilised filter should be verified by testing before use unless specifically justified and validated, and should be verified by on line testing immediately after use. Nominal pore sizes of 0.22 µm or less are acceptable without further justification, in accordance with Ph. Eur.
除非被特殊论证和验证过,除菌过滤器的完整性应在使用前通过测试进行确认, 并应在使用后立即通过在线测试进行确认。根据 Ph. Eur., 在没有进一步理由的情况下, 正常的0.22μm 或以下的孔径是可以接受的。
For routine commercial manufacturing, bioburden testing should be performed on the bulk solution immediately before sterile filtration.
对于日常的商业生产, 应在除菌过滤前立即对溶液进行生物负荷测试。
In most situations, a limit of NMT 10 CFU/100 ml (TAMC) would be acceptable for bioburden testing. If a pre-filter is added as a precaution only and not because the unfiltered bulk solution has a higher bioburden, this limit is applicable also before the pre-filter and is strongly recommended from a GMP point of view. A bioburden limit of higher than 10 CFU/100 ml before pre-filtration may be acceptable if this is due to starting material known to have inherent microbial contamination. In such cases, it should be demonstrated that the first filter is capable of achieving a bioburden of NMT 10 CFU/100 ml prior to the last filtration. Bioburden should be tested in a bulk sample of 100 ml in order to ensure the sensitivity of the method. Other testing regimes to control bioburden at the defined level should be justified.
在大多数情况下, 生物负荷测试可以接受≤10 CFU/100 ml (总需氧菌计数) 的限度。如果添加预过滤器仅为预防措施, 而不是因为待过滤溶液生物负荷过高,则此限度也适用于预过滤器之前,并且从GMP角度考虑也是强烈建议如此。如果由于起始原料已知具有固有微生物污染, 在预过滤前生物负荷限度高于10 CFU/100 ml 也是可以接受的。在这种情况下, 应证明经过第一步过滤器后在最后一个过滤器之前能够达到≤10 CFU/100 ml 的生物负荷。为了确保方法的灵敏性,生物负荷应该对100毫升的溶液进行检测。其他用以控制生物负荷在规定水平的测试方法应进行论证。
【海螺研习社解读】
这里的规定解决了长期以来的关于一级过滤器和二级过滤器接受限度的讨论和争议。从目标属性出发来解释是最好的理解的,因为生物负荷过高来通过一级二级过滤器降低本身就是悖论,因为过滤器并没有杀灭微生物本身,大量生物负荷的存在只会给过滤器本身带来更大的负担,以及使用期内的微生物风险。至于生物负荷本身的限度标准的科学性,本指南也没有进行解释,更多的情况下,最安全的做法就是根据批量、过滤器型号规格、产品特性、过滤速度、压力、甚至过滤温度来综合的评价和计算最为安全的限度。当然关于取样量,本指南强调了100毫升的底线。但是,一样这个100毫升也没有任何的科学基础。样品量和样品代表性的问题需要公司自己根据实际情况进行系统评价,批量、过程时间、溶液的物理化学状态、统计学样本量都是需要考虑的。
The maximum time between the start of bulk solution preparation and sterile filtration should be stated, minimised and appropriately supported by data. Filtration times longer than 24 hours should be justified.
应说明和最大限度减少从溶液配制到除菌过滤之间的最长时间间隔,并有数据支持。过滤时间超过24小时应进行论证。
If a sterile filtered bulk solution is not filled into the final product containers within 24 hours, the sterile filtration should, unless justified, be repeated immediately before filling. An additional bioburden test should be performed before any further bioburden reduction step after the holding time. The holding time should be adequately justified.
如果除菌过滤后的溶液未在24小时内分装入最终产品容器, 除非经论证, 否则应立即重新进行除菌过滤才能灌装。一旦超出保存时间,在任何进一步减少生物负荷的步骤之前,应进行额外的生物负荷检验。保存时间应进行充分论证。
【海螺研习社解读】
24小时的科学基础显然是微生物是活体的,保存时间过长,不可避免发生生物负荷的增加,除非有科学的数据证明其变化的趋势。当然,大家也都知道实际微生物的生长条件是受到温度、细菌种类、溶液组分、储罐条件等环境因素影响的。所以一样,24小时本身没有太科学的依据,所以不希望实际操作中大家把24小时这个限度做为金标准,金标准还是数据。
4.1.6. Aseptic processing
无菌工艺
Aseptic processing is not considered to be a sterilisation process but concerns the usage of technologies to process sterile components avoiding addition of microbiological contaminants, e.g. use of an isolator or Restricted Access Barrier System (RABS).
无菌工艺不被认为是灭菌过程, 但涉及使用对无菌组分进行处理的技术,以避免引入微生物污染,例如使用隔离器或隔离系统 (RABS)。
For aseptic processing, information on the bulk holding time before filling and on the filling time should be stated and appropriately supported by data. The times should be minimised. The grounds for holding and filling times longer than 24 hours should be justified and supported by a risk assessment. It should be verified that the results of the media simulations support the proposed holding and processing times. The actual results of media simulations fall within the field of GMP and need not be presented routinely, but may be requested by the competent authorities in certain circumstances since such data are important to justify proposed holding and filling times.
对于无菌工艺, 应说明灌装前的保存时间和灌装时长的信息, 并有适当的数据支持。时间应尽量减少。保存时间和灌装时间超过24小时的理由应经过论证并有风险评估支持。应确认培养基模拟灌装的结果能够支持保存时间和灌装时间。培养基模拟灌装的实际结果属于GMP领域,不需要例行提出,但在某些情况下, 主管当局可能会提出要求, 因为这类数据对于证明拟议的保存时间和灌装时长的合理性是很重要的。
Sterile containers should be used for aseptically treated active substances, excipients and finished products.
经无菌处理的活性物质、辅料和成品应使用无菌容器。
Where blow-fill-seal technology is used for aseptically treated products, a summary of the validation data should be provided to confirm that the container produced is sterile. The validation should, using a biological indicator with a suitable resistance, demonstrate a SAL of ≤10-6 for the surface of the container. The bioburden of the material(s) used for the manufacture of the blow-fill-seal container should be controlled. The limit should be justified in relation to the lethality of the validated blow-fill-seal process. The bioburden limit should also include a safety margin as a for any possible bioburden enclosed within the material.
当吹-灌-封技术用于在无菌处理产品时,应提供验证数据总结, 以确认所使用的容器是无菌的。验证应使用具有适当耐热性的生物指示剂, 证明容器表面的 SAL≤10-6。应控制用于制造吹-灌-封容器的材料的生物负荷。生物负荷的限度应结合吹-灌-封工艺的杀灭力进行论证。生物负荷限度还应包括安全系数,以防止材料中包含的任何可能的生物负荷。
The majority of ATMPs cannot be terminally sterilised. In such cases, the manufacturing process should be conducted aseptically. Further details on aseptic manufacturing for ATMPs can be found in the Guidelines on Good Manufacturing Practice for Advanced Therapy Medicinal Products.
大多数ATMP不能进行最终灭菌。在这种情况下, 生产过程应无菌。有关 ATMP无菌生产的更多详细信息,请参见“先进治疗药品良好生产规范指南”。
4.2. Good manufacturing practice for sterile active substances, sterile excipients and sterile containers
无菌活性物质、无菌辅料和无菌容器GMP
Volume 4 of "The rules governing medicinal products in the European Union" contains guidance for the interpretation of the principles and guidelines of good manufacturing practices for medicinal products for human and veterinary use laid down in Commission Directives 91/356/EEC, as amended by Directive 2003/94/EC, and 91/412/EEC respectively. For Advanced Therapy Medicinal Products, the Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products should be followed.
“欧盟药品管理法”第4卷包含了经第2003/94/EC号指令和第91/412/EEC号指令修正的第91/356/EEC号委员会指令中规定的人用和兽用医药产品GMP的原则和准则的解释指南。对于先进治疗药品, 应遵循“先进治疗药品的良好生产规范指南”。
4.2.1. Active substances
活性成分
The basic GMP requirements for active substances used as starting materials (European Union (EU) GMP guide part II) apply to the manufacture of sterile active substances up to the point immediately prior to the active substance being rendered sterile. The sterilisation and aseptic processing of sterile active substances are not covered by EU GMP Part II but should be performed in accordance with the principles and guidelines of GMP as laid out in the relevant EU Directive and interpreted in the GMP Guide including its Annex 1.
作为起始物料的活性物质的基本GMP要求 (欧盟 (EU) GMP指南 第二部分 ) 适用于无菌活性物质在进入无菌步骤之前的生产。无菌活性成分的灭菌和无菌处理不在欧盟 GMP 第二部分的范围内, 但应按照欧盟相关指令中的规定和 GMP指南中解释的GMP 原则(包括其附录1)和指南进行。
The sterilisation and aseptic processing of active substances is considered to be a step in the manufacture of the medicinal product. This implies that for any active substance manufacturer who performs sterilisation and subsequent aseptic handling of the active substance, a valid manufacturing authorisation or GMP certificate from an EEA authority or from an authority of countries where mutual recognition or other Community arrangements apply has to be submitted.
活性物质的灭菌和无菌加工被认为是药品生产的一个步骤。这意味着,对活性物质进行灭菌及随后无菌处理的任何活性物质生产商,需要提交EEA药监局,或互认的国家药监局颁发的或社会团体安排申请的有效的生产许可或GMP证书。
The same GMP and data requirements also apply to sterile active substances supported by a Certificate of Suitability issued by the European Directorate for the Quality of Medicines & HealthCare (EDQM) or described in an Active Substance Master File (ASMF).
同样的GMP 和数据要求也适用于由EDQM颁发的符合性证书(COS)支持的无菌活性物质, 或在活性物质主文件 (ASMF) 中描述的无菌活性物质。
4.2.2. Excipients
辅料
All the excipient sterilisation sites should be stated by name and address in the dossier.
所有的辅料灭菌工厂都应在注册文件中注明名称和地址。
For excipients required to be sterile (i.e. those subsequently used in an aseptic manufacturing process), the site where sterilisation of the excipients takes place may not have undergone inspection by an EU authority and consequently may not hold an EU GMP certificate in relation to this activity. Nevertheless the sterilisation of an excipient is a critical process and the sterility of the excipient is a critical quality attribute to ensure the sterility of the finished product. When a GMP certificate is not available, a statement should be provided confirming that the finished product manufacturer has evaluated all the manufacturers of sterile excipients with regards to their quality system related to the sterilisation of the excipient. For products for human use this evaluation should be conducted in line with the (GMP) Guidelines of 19 March 2015 on the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use by taking into account the specific requirements of Annex 1 of EU GMP-Guidelines.
对于需要无菌的辅料 (例如,直接用于无菌生产过程的辅料), 对辅料进行灭菌的工厂可能没有经过欧盟当局的检查, 因此可能无法持有与此活动相关的欧盟GMP证书。然而, 辅料的灭菌是一个关键的过程, 辅料的无菌性是确保成品无菌的一个关键的质量属性。在没有GMP证书的情况下, 应提供一份声明, 确认成品生产商已对所有无菌辅料生产商进行了与辅料灭菌相关的质量体系评价。对于人用药品,这一评价应按照2015年3月19日发布的关于正式风险评估的 (GMP) 指南进行,考虑欧盟GMP附录1的具体要求,以确定所适用的人用药品辅料的GMP。
4.2.3. Containers
容器
For containers required to be sterile (i.e. those subsequently used in an aseptic manufacturing process), the site where sterilisation of the containers takes place may not have undergone inspection by an EU authority and consequently may not hold an EU GMP certificate in relation to this activity1. When a GMP certificate is not available, certification that the sterilisation has been conducted and validated in accordance with the following ISO standards would be considered sufficient to provide an acceptable level of sterility assurance for the empty container:
对于需要无菌的容器 (例如直接用于无菌生产的容器),对容器进行灭菌的工厂可能没有经过欧盟当局的检查, 因此可能没有持有与此活动相关的欧盟GMP证书。在没有GMP证书的情况下, 证明已按照以下 ISO标准进行灭菌和验证,将被视为足以提供适当无菌保证水平的容器:
1.I.S. EN ISO 20857 Sterilization of Health Care Products - dry Heat - Requirements for the Development, Validation and Routine Control of a Sterilization Process for Medical Devices;
2.I.S. EN ISO 11135 Sterilization of Health-care Products - Ethylene Oxide - Requirements for the Development, Validation and Routine Control of a Sterilization Process for Medical Devices;
3.I.S. EN ISO 17665-1 Sterilization of Health Care Products - Moist Heat - Part 1: Requirements for the Development, Validation and Routine Control of a Sterilization Process for Medical Devices, and, ISO/TS 17665-2 Sterilization of health care products -- Moist heat -- Part 2: Guidance on the application of ISO 17665-1;
4.I.S. EN ISO 11137-1 Sterilization of Health Care Products - Radiation - Part 1: Requirements for Development, Validation and Routine Control of a Sterilization Process for Medical Devices;
5.I.S. EN ISO 11137-2 Sterilization of Health Care Products - Radiation - Part 2: Establishing the Sterilization Dose;
6.I.S. EN ISO 11137-3 Sterilization of Health Care Products - Radiation - Part 3: Guidance on Dosimetric Aspects.
It is the responsibility of the manufacturer of the medicinal product, to ensure the quality, including sterility assurance, of containers. The site where QP certification of the finished product takes place, and other manufacturing sites which are responsible for outsourcing this sterilisation activity, should have access to the necessary information to demonstrate the ongoing qualification status of suppliers of this sterilisation service. This may be checked during inspections of the manufacturer of the finished product. The Competent Authorities may also decide, based on risk, to carry out their own inspections at the sites where such sterilisation activities take place.
确保容器的质量,包括无菌保证是药品生产商的责任。对成品进行QP的工厂,以及负责将这一灭菌活动外包的其他制造工厂, 应能获得必要的信息, 以证明此灭菌服务供应商的持续确认状态。这可能会在成品生产商的检查过程中进行检查。主管当局还可根据风险决定对进行此类灭菌活动的工厂进行自己的检查。
Quality Dossier requirements
质量档案要求
The following details regarding the sterilisation of the container components should be included in the quality dossier:
关于容器部件灭菌的以下细节应包括在质量档案中:
1.The sterilisation method and sterilisation cycle;
灭菌方法和灭菌周期
2.Validation of the sterilisation cycle if the sterilisation cycle does not use the reference conditions stated in the Ph. Eur.;
灭菌周期的验证,如果灭菌周期不使用 Ph. Eur 中规定的参考条件;
3.The name and address of the site of sterilisation and, where available*, details of GMP certification of the site.
灭菌场地的名称和地址, 以及该场地 GMP 证书的详细信息(如适用) *。
*Where the container component is a CE-marked Class Is sterile device (e.g. sterile syringe), a declaration from the device manufacturer that the component is a Class Is sterile device, together with a copy of the certificate of conformity from the Notified Body will suffice. In the absence of a GMP certificate or declaration that the component is a CE-marked Class Is medical device, confirmation by finished product manufacturer that the sterilisation process has been conducted and validated in accordance with the relevant ISO standards should be provided.
* 如果容器组件是 CE标志等级I的无菌器械 (例如无菌注射器),则一份来自该器械生产商声明该组件是等级I无菌器械的声明,以及认证机构提供的符合性证书副本,足够。在没有 GMP 证书或声明组件是CE标志等级I 医疗器械的情况下, 应提供成品制造商确认已按照相关 ISO 标准灭菌和验证的证明。
【海螺研习社解读】
这部分内容要求是无可非议的,国内很多企业基于客户的要求,会去做无菌原料药出口,从本指南可以清楚的看出,实际无菌原料药的要求和无菌制剂的要求就无菌保证这部分已经没有任何的区别了,一个做原料药的企业,要承担和无菌制剂企业一样的GMP体系维护成本去满足客户的要求,那么做老板的要好好考量下客户出的价格是否真的可以涵盖这部分的成本了。
对于辅料和容器而言,更多的是提醒那些计划进行无菌制剂出口的企业,对自己的这些物料的供应商的监管和现场审计的标准。目前国内企业供应链的管理还没有提升到欧美标准的水平,既没有合格的专人也没有足够的资源配制,而国内供应商的实际状态也不能符合欧美GMP的基本要求,所以这个部分又成了我们的软肋。
4.3. Selection of sterilisation method
灭菌方法的选择
Finished products intended to be sterile should be terminally sterilised in their final container whenever possible, as clearly stated in the Ph. Eur., general chapter 5.1.1. Similarly, active substances, excipients and containers when required to be sterile should be packed before they are sterilised whenever possible. When terminal sterilisation by heat is not possible, the application of an alternative method of terminal sterilisation, sterilising filtration and/or aseptic processing may be considered. It is recognised that terminal sterilisation processes utilising conditions other than the Ph. Eur. reference conditions may be developed to provide satisfactory SALs and such alternative processes may be acceptable when properly designed, validated and controlled.
欧洲药典通论5.1.1 中明确指出, 无菌成品应尽可能在其最终容器中进行最终灭菌。同样, 需要无菌时, 活性物质、辅料和容器应包装好,然后尽可能进行灭菌。当不能用热进行终端灭菌时, 可以考虑采用一种替代终端灭菌的方法,如除菌过滤和/或无菌工艺。开发使用欧洲药典参考条件以外的其他终端灭菌工艺,以提供令人满意的SAL也是被认可的,这种替代工艺如经适当设计、验证和控制是可以接受的。
If a sterilisation process using principles other than those described in the Ph. Eur. (steam, dry heat, ionising radiation, gas sterilisation and sterilising filtration) is intended to be used for the sterilisation of an active substance, excipient, container or finished product, the applicant may consider seeking scientific advice regarding the acceptability of the method and the documentation required.
如果打算使用欧洲药典中(蒸汽、干热、电离辐射、气体灭菌和除菌过滤)以外的其他灭菌方法对活性物质、辅料、容器或成品进行灭菌,申请人可考虑就该方法的可接受性和所需文件寻求科学意见。
During the manufacturer’s evaluation of whether a terminal sterilisation cycle is possible, substantial efforts should be made to enable terminal sterilisation. If the active substance or another component of the finished product is shown to degrade significantly or an impurity limit is exceeded during shelf-life under even the least stressful terminal sterilisation conditions, the efforts made to develop a formulation and container capable of undergoing terminal sterilisation should be presented in the development section. Such efforts could be selection of optimal pH, choice of excipients (qualitative and quantitative), container, optimisation of sterilisation method and manufacturing conditions.
在生产商评估是否有可能进行最终灭菌的过程中,应尽可能使用终端灭菌。如果即使在最缓和的最终灭菌条件下, 活性物质或成品的其他成分在其有效期内仍会明显下降或杂质超标,则应在研发部分应说为开发耐受最终灭菌工艺的配方和容器所做的努力,包括选择最佳 ph 值、选择辅料 (定性和定量)、容器、优化灭菌方法和生产条件。
In case of medicinal products containing highly sensitive active substances, (e.g. proteins or other heat labile biological substance), where it is well known that terminal sterilisation is not possible, a justification based on a scientific rationale is generally acceptable and further justification of the choice of aseptic processing discussed later in section 4.3 may not be needed.
如果是含有高敏感性的活性物质 (如蛋白质或其他热不稳定的生物物质) 的医药产品, 众所周知, 不可能进行最终灭菌, 基于科学合理的论证通常是可以接受的,不需要进一步论证选择第4.3 节无菌工艺的理由。
The principles for the choice of sterilisation process for finished products and containers are presented in the form of decision trees in section 5 of this guideline. The principles of the decision trees may also be applied for the sterilisation of active substances and excipients.
本指南第5节以决策树的形式介绍了成品和容器灭菌工艺选择的原则。决策树的原则也可适用于活性物质和辅料的灭菌。
For finished products where terminal sterilisation is not possible and aseptic processing is proposed, the decision trees should be applied to individual components or mixtures of components in the formulation. An impact on the shelf-life or storage conditions caused by a terminal sterilisation process is not in itself a reason to exclude terminal sterilisation, unless the new storage condition or shelf-life would cause significant problems for the user.
对于不可能进行最终灭菌并拟进行无菌处理的成品,决策树应应用于配方中的单个组分或组分的混合物。最终灭菌过程对有效期或储存条件造成的影响本身并不是排除最终灭菌的理由, 除非新的储存条件或有效期会给用户带来重大问题。
Terminal sterilisation should not be ruled out purely on the basis of an increase in degradation products above the qualification thresholds in ICH Q3A/VICHGL10 (active substances), ICH Q3B/ VICH GL11 (finished products) or the impurity limits in ICH M7 for products in the scope of that guideline without additional justification. If impurities are either metabolites or are generated at levels already qualified, then terminal sterilisation is still considered feasible. However, if the degradation products are not qualified at the level at which they occur, then sterile filtration and aseptic processing may be selected. For medicinal products for human use impurities which occur above the identification threshold should be specified in the finished product specification.
不应仅仅因为降解产物增加超过 ICH Q3A/VICHGL10 (活性物质)、ICH Q3B/VICH GL11 (成品) 中的限度或ICH M7指导范围中的产品的杂质限度,就排除最终灭菌而不进行论证。如果杂质都是代谢产物或是产生的杂质在确认水平, 那么最终灭菌仍然被认为是可行的。然而, 如果产生的降解产物的水平不在其确认水平, 则可以选择除菌过滤和无菌工艺。对于人用药产品, 超过鉴别阈值的杂质应在成品标准中明确规定。
The risk induced by the degradation should be balanced by the risk induced with an aseptic manufacturing method, also taking in account the posology of the finished product and the nature of the degradation products. Attempts to find terminal sterilisation conditions adjusted to give acceptable impurity levels based on degradation mechanisms of the active substance and the actual bioburden should be described in the quality dossier.
降解引入的风险应与无菌生产方法引入的风险进行平衡, 同时考虑到成品的剂量和降解产物的性质。在质量档案中, 应说明根据活性物质的降解机制和实际生物负荷,调整最终灭菌条件,得到可接受的杂质水平而进行的尝试。
【海螺研习社解读】
这部分更加实际的解释了在选择“最终灭菌”还是“无菌工艺”的区别和思考点,尤其是对降解杂质的限度考量给出了允许超出相关指南的条件。这里的灵活性基于的原则还是已知降解杂质的风险大小和无菌性能的风险大小的权衡。这个思考点和优先等级的处置,值得我们关注和在技术开发的过程中先期导入。
In certain cases, as described in the bullet points below, the use of aseptic processing may be accepted, even if the formulation itself can be terminally sterilised. The approach should be clearly documented, explained and scientifically justified. Such cases could be justified by:
在某些情况下, 如下文要点所述, 即使配方本身可以最终灭菌, 也可以使用无菌工艺。应明确记录、解释和科学论证这一方法。这种情况的通过以下方式论证:
User benefit provided by a container that cannot be terminally sterilised such as:
不能最终灭菌的容器有利于用户利益, 例如:
Eye drop containers enabling administration of single drops to the eye;
能实现单滴液滴滴到眼睛里的滴眼容器;
Containers enabling non parenteral multi-dose preservative free medicinal products for human use;
能实现非注射用多剂量无防腐剂的人用药品的容器;
Enhanced ease of administration;
更易服用;
Safer handling of toxic products, for instance plastic vials instead of glass vials for cytotoxic medicinal products.
更安全地处理有毒药品,例如细胞毒性药品使用塑料瓶而不是玻璃瓶。
The choice to use a heat-labile container cannot in itself be the sole reason for not applying a terminal sterilisation process and alternative materials should be investigated. Thus, a discussion regarding the efforts made to develop a container that may be terminally sterilised should be included.
选择使用热不稳定容器本身不能成为不采用最终灭菌工艺的唯一理由,应研究使用替代材料。因此,应包含关于研究可以最终灭菌的容器的研究的讨论。
Enabling as long a shelf-life as possible for radiopharmaceutical medicinal products with a shelf-life of less than one week.
为有效期少于一周的放射性药物产品提供尽可能长的保质期。
【海螺研习社解读】
这部分的两个情形,天平又向无菌工艺倾斜,原因是显而易见的,微生物的潜在危害是可控的,是和产品的效应和病人的其他应用危害进行对比的选择。
The acceptability of aseptic processing should be based on the application of the decision tree and a risk assessment. The bullet points below are not intended to be used to justify aseptic processing as such, but are only intended to provide guidance on issues that are considered when evaluating the acceptability of a sterilisation or aseptic processing. Considerations include (but are not limited to):
无菌工艺的可接受性应基于决策树的应用和风险评估。下面的要点并非旨在用于证明无菌工艺的合理性,而仅旨在为评估灭菌或无菌工艺的可接受性时需考虑的问题提供指导。考虑因素包括(但不限于):
Evidence that the proposed container with enhanced user benefits is fit for purpose;
推荐的容器可以提高用户利益的证据;
Stability of the active substance, the degradation mechanism(s) and the toxicity of impurities formed during the sterilisation process;
活性物质、灭菌过程中产生的杂质的降解机理和毒性的稳定性;
The volume to be administered per dose.
每剂给药的体积
In conclusion, the justification for the chosen sterilisation or aseptic processing should include a thorough benefit risk evaluation and it should be demonstrated that suitable development efforts have been made.
总之,所选择的灭菌或无菌工艺的理由应包括详细的利益风险评估,并且应该证明已经进行了适当的研发工作。
For advanced therapy medicinal products, the microbiological quality of all components, process equipment and the aseptic techniques of the manufacturing processes are of utmost importance when the finished product cannot be sterilised. For those medicinal products that cannot be sterilised, such as cell based medicinal products, a detailed risk assessment with regards to microbial contamination should be provided. A risk based approach is already foreseen for these ATMP (see Guideline on the risk-based approach according to annex I, part IV of Directive 2001/83/EC applied to Advanced therapy medicinal products, EMA/CAT/CPWP/ 686637/2011).
对于先进的治疗药品,当成品不能灭菌时,所有组分的微生物学质量,工艺设备和生产工艺的无菌技术都是至关重要的。对于那些不能灭菌的药品,如基于细胞的药品,应提供有关微生物污染的详细风险评估。对这些ATMP产品,基于风险的方法已经被预见到。(参见基于风险的方法指南,根据适用于高级治疗药物的指令2001/83 / EC的第IV部分附录I,EMA / CAT / CPWP / 686637/2011)。
5. Decision trees
决策树
The decision trees in Figures 1 and 2 are intended to assist in the selection of the optimal sterilisation method taking into account the various issues to be considered. When moving down the decision trees, the methods generally show a decreasing assurance of sterility and therefore, the first feasible option should normally be chosen.* The decision trees have been elaborated primarily for finished products containing chemical active substances, but may be applicable also to other types of products (including active substance and excipients). Figure 3 provides the corresponding information for empty containers. The decision tree is not applicable to sterile empty containers that are CE marked medical devices. In the case of biological products, an alternative approach may be appropriate.
图1和图2中的决策树旨在帮助选择最佳的灭菌方法,同时考虑到需要考虑的各种问题。决策树越向下的方法,无菌保证越低, 因此, 通常应选择第一个可行的选择*. 决策树主要针对含有化学活性物质的成品进行了详细阐述,但也可适用于其他类型的产品(包括活性物质和辅料)。图3提供了空容器的相应信息。决策树不适用于有CE标记的医疗器械的无菌空容器。对于生物产品,可采用替代办法。
*While sterilisation by heat and sterilisation by ionising irradiation provide the same assurance of sterility, sterilisation by heat has lower risk (e.g. radiolysis impurities) and is more easily controlled than sterilisation by ionising irradiation. For these reasons, heat is given priority over ionising irradiation in the decision trees.
虽然通过加热灭菌和通过电离辐射灭菌提供了同样的无菌保证, 但加热灭菌的风险低(辐射杂质更加复杂),也比电离辐射灭菌更容易控制。因此, 在决策树中, 加热灭菌优先于电离辐射灭菌。
【海螺研习社解读】
这部分和之前没有显著性的区别,没有更多值得讨论和解读的地方。
Figure 1 Decision tree for sterilisation choices for aqueous products
图1 对于溶液剂型产品灭菌方法选择的决策树:
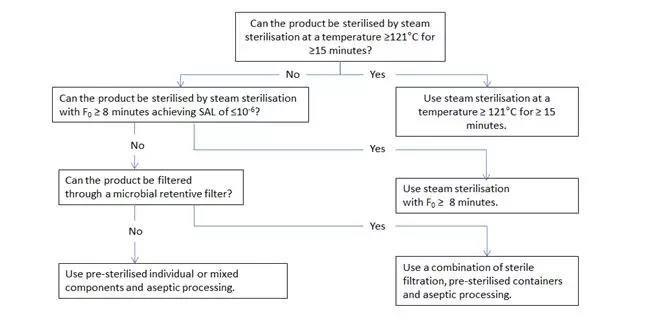
Figure 2 Decision tree for sterilization choices for dry powder products, non-aqueous liquid or semi-solid products
图2 干粉产品,非溶液剂型和半固体产品灭菌方法选择的决策树:
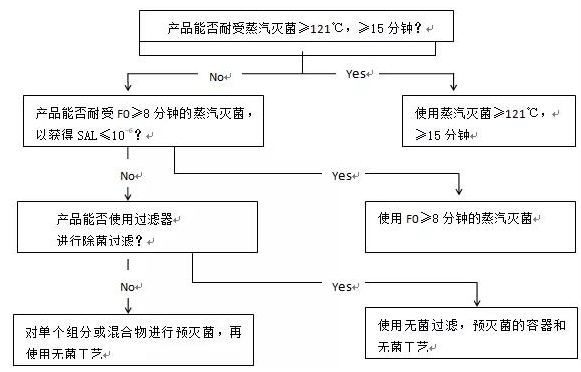
Figure 3 Decision tree for sterilization choices for containers
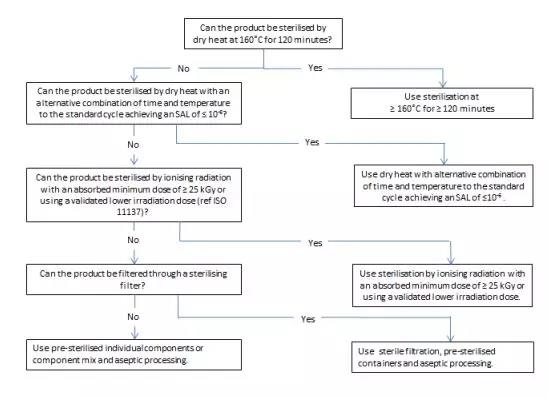
图3 容器灭菌工艺选择的决策树
6. Definitions
名词解释
Aseptic processing
无菌工艺 A process performed maintaining the sterility of a product that is assembled from components, each of which has been sterilised by steam, dry heat, ionizing radiation, gas or sterile filtration. This is achieved by using conditions and facilities designed to prevent microbiological contaminants.
一种用于保持由各种经过蒸汽、干热、电离辐射、气体或除菌过滤的方式进行了灭菌的部件组合而成的产品的无菌性的工艺。这种工艺是通过使用旨在防止微生物污染物的条件和设施来实现的。
Bioburden
微生物负荷(负荷) The total number of micro-organisms associated with a specific item prior to any sterilisation or bioburden reduction step.
在任何灭菌或生物减少步骤之前与特定物品相关的微生物总数。
Biological indicator
生物指示剂 Biological indicators are test systems containing viable microorganisms (usually spores of bacteria) that provide a defined challenge to verify the required effectiveness of a specified sterilisation process.
生物指标是包含活的微生物 (通常是细菌孢子) 的测试系统, 用来验证特定灭菌过程具备所需的有效性的挑战。
Colony Forming Unit (CFU)
菌落数 A microbiological term that describes the formation of a single macroscopic colony after the introduction of one or more micro-organisms to microbiological growth media. One colony forming unit is expressed as 1 CFU.
一个微生物术语,用来描述在微生物培养基上加入一个或多个微生物后形成的肉眼可见的菌落数。一个菌落形成单位标示为1 CFU.
Critical Quality Attribute
关键质量属性 A physical, chemical, biological or microbiological property or characteristic that should be within an appropriate acceptance criteria, range, or distribution to ensure the desired product quality.
应符合一定接受标准、范围或分布的物理、化学、生物或微生物属性或特性,用来确保目标产品质量。
Depyrogenation
除热源 A process used to destroy or remove pyrogens (e.g. endotoxins).
用来破坏或去除热源(或内毒素)的过程。
D-value (decimal reduction value)
D值(十进制减少值) The value of a parameter of sterilisation (duration or absorbed dose) required to reduce the number of viable organisms to 10 per cent of the original number. It is only of significance under precisely defined experimental conditions. D121 is the D-value of the relevant spores at 121C.
将活生物体数量减少到原数量的10%所需的灭菌参数(持续时间或吸收剂量)的值。只有在精确定义的实验条件下,才有意义。D121是在121C时相关孢子的D值。
F0 value
F0值 The F0 value of a saturated steam sterilisation process is the lethality expressed in terms of the equivalent time in minutes at a temperature of 121 °C delivered by the process to the load in its container with reference to micro-organisms possessing a theoretical Z-value of 10.
饱和蒸汽灭菌过程的 F0 值是指对容器中的负载在121°C时进行灭菌时, 参照理论Z 值为10的微生物表示的灭菌效果的等效时间,以分钟为单位。
Filling time
灌装时间 The time used to fill a bulk product into containers until the container is closed or, in the case of a product which is lyophilized after the filling, until the lyophilisation chamber is closed.
将散装产品灌装入容器到容器封闭的时间, 或者, 如果是在灌装后进行冻干的产品, 到冻干室关闭的时间。
Holding time
保存时间 The time between two process steps.
两个工艺步骤之间的时间。
Immunological veterinary medicinal product
免疫学兽药产品 A veterinary medicinal product administered to animals in order to produce active or passive immunity or to diagnose the state of immunity.
一种用于动物的兽药产品, 使动物产生主动或被动免疫或诊断免疫。
Lethal (process)
致死 A process that kills the microorganisms exponentially.
杀死微生物的过程。
Overkill sterilisation
过度灭杀
A process with a lethality of F0BIO > 12 minutes. For example a process that provides at least a 12 log reduction of biological indicator micro¬organisms having a minimum D value of 1 minute.
具有F0BIO > 12 分钟灭杀力的工艺。比如,能够提供至少12log微生物降低的最小 D 值为1分钟。
Ph. Eur. sterilisation reference conditions
欧洲药典参考灭菌条件 The reference conditions for sterilisation specified in Ph. Eur. 5.1.1, i.e. terminal steam sterilisation at ≥121 °C for 15 min, terminal dry heat sterilisation at ≥160 °C for ≥2 h or terminal ionising radiation of 25 kGy.
Ph.Eur.5.1.1中列出的参考灭菌条件,比如终端蒸汽灭菌≥121 °C ,持续15 min,终端干热灭菌≥160 °C,持续≥2 h,或者终端电离辐射灭菌25 kGy。
Post-aseptic processing terminal heat treatment
无菌工艺后进行终端加热处理 A terminal moist heat process employed after aseptic processing which has been demonstrated to provide a SAL ≤10-6, but where the requirements of steam sterilisation (for example, F0≥8 min) are not fulfilled.
在已证明可以提供SAL ≤10-6的无菌保证水平的无菌工艺后进行的终端湿热工艺,但是不满足对蒸汽灭菌的要求(比如F0≥8 分钟)。
SAL
无菌保证水平 Sterility Assurance Level. The SAL for a given sterilisation process is expressed as the probability of micro-organisms surviving in a product item after exposure to the process. An SAL of 10-6, for example, denotes a probability of not more than 1 non-sterile item in 1 × 106 sterilised items of the final product.
无菌保证水平。对于给定的灭菌过程来讲,无菌保证水平指的是微生物在经历了灭菌过程后的存活率。比如SAL=10-6, 表示当有1 × 106个最终产品进行灭菌后,有不超过1个有可能有菌。
Slowest to heat locations
最慢加热位置 Location in the load that remains coldest or where the temperature is raising slowest during the sterilisation process.
It could, in a figurative sense, also be used for other sterilisation methods for the location in the load achieving the lowest level of sterilising energy.
装载中的最冷点的位置或者在灭菌过程中温度上升最慢的位置。
也可以比喻为表示在其他灭菌方法中在负载中获得的灭菌能量水平最低的位置。
Steam sterilization
蒸汽灭菌 Reference is made to the description in Ph. Eur. 5.1.1.
参考在Ph.Eur.5.1.1中的描述。
Sterilisation
灭菌 A suitably designed, validated and controlled process that inactivates or removes viable micro¬organisms in a product until sterility is obtained.
Sterility is the absence of viable microorganisms, as defined by a sterility assurance level equal to or less than 10−6.
The inactivation of microorganisms by physical or chemical means follows an exponential law; thus there is always a finite statistical probability that a micro-organism may survive the sterilising process. For a given process, the probability of survival is determined by the number, types and resistance of the microorganisms present and by the environment in which the organisms exist during treatment.
经过适当设计,验证和控制的过程,用来杀灭或者去除产品中的活的微生物,使产品达到无菌的过程。
像无菌保证水平等于或者小于10−6时的定义一样,无菌是指没有活的微生物。
通过物理或化学手段来杀灭微生物遵循指数规律;因此, 微生物在灭菌过程中存活的概率总是有限的。对于给定的过程, 生存的概率取决于存在的微生物的数量、类型和耐力, 以及在处理过程中所处的环境。
TAMC
总需氧菌数 Total aerobic microbial count: The total aerobic microbial count (TAMC) is considered to be equal to the number of CFU found using casein soya bean digest agar.
总需氧菌数:被认为等于使用酪蛋白大豆消化琼脂时发现的 CFU 数量。
Terminal process
终端工艺 A process where a finished product is processed in its primary container, for example terminal sterilisation or post-aseptic processing terminal heat treatment.
最终产品在内包材中被进行加工的过程, 例如终端灭菌或无菌处理终端后热处理。
Validation
验证 Establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes.
建立书面证据证明某个特定流程能够高度保证能够始终如一地生产出符合其预定规格和质量属性的产品。
Worst case
最差条件 A set of conditions encompassing upper and lower processing limits and circumstances, including those within standard operating procedures, that pose the greatest chance of process or product failure (when compared to ideal conditions). Such conditions do not necessarily induce product or process failure.
包含标准操作规程中最高和最低限度和环境的一组条件,可能会最大限度的导致工艺失败或产品不合格(当与理想提交相比时)。这些条件不一定总是会导致产品不合格或工艺失败。
z-value
Z值 The z-value is the change in temperature required to alter the D-value by a factor of 10.
Z 值是将 d 值更改10倍所需的温度变化。
7. References
Decision trees for the selection of sterilisation methods, CPMP/QWP/054/98;
Note for Guidance: Development Pharmaceutics for veterinary medicinal products: Decision tree for the selection of sterilisation methods, EMEA/CVMP/065/99;
Note for guidance on manufacture of the finished dosage form, CPMP/QWP/486/95;
Note for Guidance: Manufacture of the finished dosage form, EMEA/CVMP/126/95;
ICH guideline Q8 (R2) on pharmaceutical development, EMA/CHMP/ICH/167058/2004;
European Pharmacopoeia general chapter 5.1.1 ‘Methods of preparation of sterile products’;
Note for Guidance: Virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses, EMA/CPMP/BWP/268/95;
Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use, as amended;
Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products, as amended;
Guideline on real time release testing (formerly Guideline on parametric release), EMA/CHMP/QWP/811210/2009-Rev1;
Guideline on Parametric release, EMEA/CVMP/QWP/339588/2005;
EudraLex - Volume 4 Good manufacturing practice (GMP) Guidelines;
European Pharmacopoeia general chapter 5.1.5 ‘Application of the F0 concept to steam sterilisation of aqueous preparations’;
European Pharmacopoeia general chapter 5.1.2 ‘Biological indicators and related microbial preparations used in the manufacture of sterile products’;
NfG on The use of Ionisation Radiation in the Manufacture of Medicinal products 3AQ4A;
EN/ISO 11137, Sterilisation of health care products – Radiation;
ICH guideline M7 on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk (EMA/CHMP/ICH/83812/2013);
European Pharmacopoeia general chapter 5.1.7 ‘Viral Safety’ Human cell-based medicinal products, EMEA/CHMP/410869/2006
Questions and Answers on allogenic stem cell-based products for veterinary use: specific questions on sterility EMA/CVMP/ADVENT/751229/2016
Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products, EMA/CAT/80183/2014
I.S. EN ISO 20857 Sterilization of health care products - dry Heat - Requirements for the development, validation and routine control of a sterilization process for medical devices
I.S. EN ISO 11135 Sterilization of health-care products - Ethylene Oxide - Requirements for the development, validation and routine control of a sterilization process for medical devices I.S. EN ISO
17665-1 Sterilization of health care products - Moist heat - Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices
ISO/TS 17665-2 Sterilization of health care products -- Moist heat -- Part 2: Guidance on the application of ISO 17665-1
I.S. EN ISO 11137-1 Sterilization of health care products -Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices
I.S. EN ISO 11137-2 Sterilization of health care products -Radiation - Part 2: Establishing the sterilization dose
I.S. EN ISO 11137-3 Sterilization of health care products -Radiation - Part 3: Guidance on dosimetric aspects of development, validation and routine control
ICH Q3A (R2) Impurities in new drug substances, CPMP/ICH/2737/99
ICH Q3B (R2) Impurities in New Drug Products, CPMP/ICH/2738/99;
VICH GL10 Impurities in new veterinary drug substances, CVMP/VICH/837/99 Rev.1
VICH GL11 Guideline on impurities in new veterinary medicinal products, EMEA/CVMP/VICH/838/99 Rev.1.
Guideline on the risk-based approach according to annex I, part IV of Directive 2001/83/EC applied to Advanced therapy medicinal products, EMA/CAT/CPWP/686637/2011).
文章来源:海螺研习社
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。