扫一扫 添加小助手
服务热线
13818320332
扫一扫 关注我们

6 March 2019

本指南替代“药物研发指南”(CPMP/QWP/155/96)的附件文件“灭菌方法选择决策树”(CPMP/QWP/054/98)和“兽药研发指南”(EMEA/CVMP/315/98)的附件文件“灭菌方法选择决策树”(EMEA/CVMP/065/99)
Table of contents 目录
1. Introduction (background)概述(背景)
2. Scope 范围
3. Legal basis 法律依据
4. General requirements一般性要求
4.1. Requirements for the manufacture of sterile medicinal products and sterile components 无菌医药产品和无菌组件的生产制造要求
4.1.1. Steam sterilisation 蒸汽灭菌
4.1.2. Dry heat sterilisation 干热灭菌
4.1.3. Ionization radiation sterilisation 放射灭菌
4.1.4. Gas sterilisation 气体消毒灭菌
4.1.5. Sterile filtration 除菌过滤
4.1.6. Aseptic processing 无菌工艺
4.2. Good manufacturing practice for sterile active substances, sterile excipients and sterile containers 无菌原料药、无菌辅料和无菌包材的GMP要求
4.2.1. Active substances 原料药
4.2.2. Excipients 辅料
4.2.3. Containers 包材
4.3. Selection of sterilisation method 灭菌方法的选择
5. Decision trees 决策树
6. Definitions 术语表
7. References 参考文献
Executive summary
实施摘要
Guidance is provided on the selection of appropriate methods of sterilisation for sterile products. Although, terminal sterilisation using a reference condition of the European Pharmacopoeia (Ph. Eur.) is the method of choice whenever possible, this guideline provides information on when other terminal sterilisation processes, sterilising filtration or aseptic processing, (either alone or when combined with an additional post-aseptic processing terminal heat treatment), could be accepted as an alternative.
本指南提供无菌药品灭菌方法的选择。尽管使用参考EP条件进行终端灭菌是优先选择的方法,本指南提供了当其它终端灭菌工艺、除菌过滤或无菌工艺(单独或与无菌工艺后加终端热处理工艺联合使用)可以被接受作为替代方法时的信息。
Guidance is provided on the documentation expected for sterile finished products, sterile active substances, sterile excipients and sterile primary containers (referred to as container in this guideline) in a new marketing authorisation application or a variation application for a medicinal product, (called quality dossier throughout the guideline).
本指南提供了关于新的无菌产品上市许可申报或无菌产品变更申报(本指南中称为“质量文档”)中的关于对无菌制剂产品,无菌原料药,无菌辅料和无菌内包材(本指南中称为“包装”)的文档预期。
Terminology definitions are included at the end of the document.
相关术语的定义参考本指南末端的说明
1. Introduction (background)
概述(背景)
Sterility is a critical quality attribute for all sterile substances, products and containers. Sterility cannot be assured by testing, it needs to be assured by the use of a suitably designed, validated and controlled manufacturing process. Sterility is achieved by controlling several factors such as the bioburden, the sterilisation procedure, the integrity of the container closure system and in the case of aseptic processing, the use of satisfactory aseptic technique.
无菌是所有无菌原料药、药品和包材的关键质量属性。药品的无菌不能通过测试来保证,它需要由使用适当设计的、经过验证的且受控的生产工艺来确保。无菌控制可以通过多个因素来实现,例如生物负荷、灭菌程序、容器密闭系统的完整性、对于无菌工艺,则是取决于满意的无菌技术的应用。
Terminal sterilisation is preferred to sterilisation by filtration and/or aseptic processing because it is lethal to micro-organisms and a reliable sterility assurance level (SAL) is possible to calculate, validate and control, and thus incorporates a safety margin. For sterile filtration followed by aseptic processing, this is not applicable as accidental contamination caused by inadequate technique cannot be reliably eliminated by monitoring and control. Therefore, terminal sterilisation provides the highest assurance of sterility and should be used whenever possible.
终端灭菌永远优先于除菌过滤和/或无菌工艺,因为它能消灭微生物且可以提供可以计算、验证和控制的更加可靠的无菌保证水平(SAL),从而构建了安全边际。对于采用除菌过滤后,进行的无菌工艺来说,由于技术不充分而导致的偶然污染使得其SAL不完全可靠,因为这些污染无法通过监测或控制可靠地清除。所以终端灭菌可以带来较高水平的无菌保证,必须始终优先考虑。
For highly sensitive products, such as most biological products, where terminal sterilisation of the finished product is not possible, sterile filtration and/or aseptic processing under validated and controlled conditions can be accepted.
对于高度热敏感的产品,例如生物药品,如果药品无法进行终端灭菌,在受控和验证条件下的除菌过滤和/或无菌工艺是可以被接受的。
Sterile filtration and aseptic processing are closely related and difficult to consider separately, since sterile filtration in most cases is followed by at least one aseptic processing step such as filling. In order to focus on the most important aspect of filtration and aseptic processing at each section of this guideline, only one of the two steps may be mentioned, even if both steps are related.
除菌过滤和无菌工艺是高度相关很难区分的,因为除菌过滤在大多数情况下都会涉及到至少一种无菌工艺步骤,例如无菌灌装。本指南为了集中关注最重要的除菌过滤和无菌工艺,可能仅仅会提到其中的一项技术,即使两者实际是关联的。
In addition to those finished products where the formulation itself prohibits the possibility of terminal sterilisation, the use of aseptic processing can be accepted in certain situations, even if the formulation itself can be terminally sterilised, if other benefits are gained for patients or users of the product. These situations are specified below in section 4.3.
另外,对于那些配方本身使得终端灭菌无法进行的那些药品(即使配方本身可以进行最终灭菌,如果患者或药品用户可以从无菌工艺中获得其它好处),使用无菌工艺在特定的情形下也是可以接受的。这些情形在以下第4.3部分描述。
Container integrity is discussed in ICH Q8, (adopted for human medicinal products only, nevertheless the same principles are also applicable to veterinary medicinal products and containers of sterile substances and containers).
容器完整性在ICH Q8里进行了讨论(正式采纳仅适用于人用药品,但是相同的原则还是适用于兽药)。
2. Scope
范围
The guideline applies to chemical and biological medicinal products for human and veterinary use but is not applicable to immunological veterinary medicinal products.
本指南适用于人药和兽药的化学和生物制剂,但不适用于兽用免疫制品。
It is acknowledged that the recommendations provided for in this guideline may require some adaptation to the specific characteristics of Advanced Therapy Medicinal Products (ATMPs) for human use (e.g. difficulties to differentiate between starting material, active substance and finished product in some cases, scarcity of starting materials/active substance/finished product (autologous products and matched-donor scenario), small volumes of production). The level of documentation that is expected to be included in marketing authorisation applications for ATMPs may be adapted provided that this is justified under a risk-based approach. For veterinary cell based novel therapies, cross reference is made to EMA/CVMP/ADVENT/751229/2016 Questions and Answers on allogenic stem cell-based products for veterinary use: specific questions on sterility.
本指南中提供的建议可能需要对“人用先进治疗药物”(ATMPs)的特定特性进行一些调整(例如,在某些情况下难以区分起始物料、活性物质和成品、起始物料/活性物质/成品相对稀缺【自体产品和匹配供体方案】,小批量生产)。如果根据基于风险评估证明这是合理的,则可以在ATMPS上市申请中的调整所需的文档级别。对于基于兽医细胞的新疗法,交叉参考EMA/CVMP/Advent/751229/2016的“关于兽用同种干细胞产品的问答:关于无菌的具体问题”。
Guidance is provided on the choice of sterilisation method, the development data and manufacturing data required to demonstrate the suitability of the selected sterilisation process. The same principles (choice of method of sterilisation, development data and manufacturing data) apply to sterile active substances, excipients and primary containers. Only the information expected in the quality dossier, including information related to Good Manufacturing Practice (GMP) certificates, is described. Not all GMP requirements (e.g. environmental monitoring, sterilisation of manufacturing equipment) are referenced in the guideline, only those that are considered specifically relevant for the quality dossier.
本指南提供了灭菌方法的选择,在进行合适的无菌工艺选择的时候必须考虑具体产品的研发数据和生产数据。对于无菌原料药、辅料和内包材,相同的原则(选择灭菌方法、研发数据和生产数据)也一样适用。本指南只描述了对质量文档中所需的信息,包括关于GMP证书需求信息,不包括通用的GMP要求(比如环境监控,生产设备的灭菌等)。
The scope of this document includes:
本指南的范围包括:
Terminal sterilisation by steam, dry heat and ionising irradiation using the reference conditions of Ph. Eur. 5.1.1 “Methods of preparation of sterile products” or other conditions stated in that monograph
采用EP5.1.1“无菌产品的制备方法”或其他药典给定的条件,采用蒸汽、干热和放射源进行的终端灭菌的方法
Sterilisation by filtration and aseptic processing
除菌过滤和无菌工艺方法
Sterilisation by gas
使用气体进行消毒灭菌的方法
The concepts in this guideline refer only to absence or removal of bacteria, fungi and bacterial endotoxins. The absence, removal or inactivation of viruses, mycoplasma, prions and other adventitious agents, which could contaminate a product, are not considered. For virus validation reference is made to the Guideline Virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses, CPMP/BWP/268/95.
在本指南中的概念仅指细菌、霉菌和细菌内毒素的清除情形。病毒、支原体、朊病毒和其它外来可能污染药品的试剂的消除、清除或灭活不在本指南范围。对于病毒验证请参考CPMP/BWP/268/95 “病毒验证指南:病毒灭活和清除验证的设计、实现和阐述”
【海螺研习社解读】
概述很实在,反复强调了终端灭菌的地位以及其背后的科学逻辑。只有终端灭菌是真的杀灭了微生物,并且其杀灭的效果可以进行客观的控制和检测出来。其他任何的无菌工艺技术因为存在技术上的天生局限性,无法对微生物进行杀灭,无法对微生物的最终状态进行定量客观的控制以及明确的检测,所以都存在一定的理论和实际上的交叉污染的风险。任何法规背后都必须是科学基础,这点在这里体现的很好!
特殊制剂类型,特殊产品,特殊包材性质决定了不可能在实际操作中都使用终端灭菌,所以才有了本指南的讨论,也才有了无菌工艺这个说法,也才有了GMP上最难监管、控制和测试的一个领域,这个领域不可避免的引入了人这个最大的可变要素,所以我们才说在整个GMP监管上无菌工艺的难度是最难的,也是最有挑战的。
回到现实,本指南的适用范围在本章节也进行了框定。基本符合我们最熟悉的无菌和灭菌过程,没有更多进行讨论的余地。
3. Legal basis
法规依据
This guideline should be read in conjunction with Directive 2001/83/EC on the community code relating to medicinal products for human use, Directive 2001/82/EC on medicinal products for veterinary use as amended and also the current Ph. Eur.
本指南应与欧盟人用药指令2001/83/EC和欧盟兽药指令2001/82/EC及其修订内容以及现行EP一起解读。
In addition, this guideline should be read in conjunction with all other relevant directives and regulations, and all relevant Commission, (V)ICH and CXMP guidelines, Q&A documents and other documents as linked to or published on the EMA website (www.ema.europa.eu).
此外,本指南应与所有其它相关指令和法规,以及所有相关委员会、(V)ICH和CXMP指南、问答文件及其它相关文件或EMA网站(www.ema.europa.eu)公布的文件一起解读。
4. General requirements
一般性要求
The guideline concerns specific requirements related to sterility, sterilisation processes and aseptic processing of sterile products and product components.
本指南只是针对无菌产品和产品组分的无菌,灭菌工艺和无菌工艺的特定要求。对于药品生产中的其它要求,参见其它指南如制剂生产指南。
4.1. Requirements for the manufacture of sterile medicinal products and sterile components
无菌药品和无菌组件的生产制造要求
The choice of sterilisation method or aseptic processing should be justified, see section 4.3 Selection of sterilisation method.
灭菌方法或无菌工艺的选择应进行合理化解释,参见第4.3部分灭菌方法的选择。
All sterilisation processes should be carried out according to the instructions of the Ph. Eur. unless justified.
所有灭菌工艺过程应根据EP要求实施,除非另有合理理由。
【海螺研习社解读】
“Justification”是典型的欧美用语,也是和中国监管当局目前的评审态度最大的区别所在。当然,我们目前自己的业内企业也不具备“合理科学解释”的综合能力。这里需要进行解释的是两个要点:为啥要选择某个灭菌/无菌工艺?某个灭菌/无菌工艺的实施细节是否和EP规定一致,如果不一致,为什么允许其不一致?
大部分有过实际欧美注册经验的朋友,都知道在进行这类问题的阐述的时候,最好的理由不是“引经据典”,最好的理由是采用科学实验的直接的监测数据(“摆事实、列数据”)。
All sterilisation procedures for the finished product, active substance, the excipient(s) or the containers and the name and address of the sterilisation site should be stated. A description of the sterilisation method and/or aseptic processing, including in-process controls and validation data should be provided.
所有制剂成品、原料药、辅料或内包材的灭菌程序应进行描述,还必须注明负责灭菌的工厂名称和地址。应按以下所述要求提供每个灭菌工艺的灭菌方法和/或无菌工艺方法,包括中控和验证数据。
【海螺研习社解读】
在目前的市场环境下,大量的无菌操作开始被专业化的机构和公司分包,比如包材有专业的公司进行灭菌处理,比如某些特殊的灭菌工艺(例如辐射灭菌)委托给专业的机构进行操作,那么这些机构的相关信息、工艺数据、以及相关的质量协议和审计报告就不能缺席了。在实际操作中,这类委托机构,因为服务大量的不同客户,其标准往往是相对统一的,对于存在特殊要求的企业,需要在质量协议和现场监管上下功夫,确保自己的特殊要求被满足。另外,也要高度重视这类机构的合规风险,如果其客户群中存在不规范的行为,药监当局会采取措施封杀整个委托机构,那么自己的产品也会被“误伤”。
When parametric release of sterility is proposed, the Guideline on real time release testing (formerly Guideline on parametric release), EMA/CHMP/QWP/811210/2009-Rev1 (human products only), the Guideline on Parametric release, EMEA/CVMP/QWP/339588/2005 (veterinary products only) and the text of Ph. Eur. Chapter 5.1.1 should be taken into account.
如果采用无菌参数放行,应考虑实施“实时放行测试指南”(之前的“参数放行指南”) EMA/CHMP/QWP/811210 /2009-Rev1 (仅人药)和“参数放行指南”EMEA/CVMP/QWP/ 339588/2005 (仅兽药)以及EP第5.1.1章文本。
The bioburden control criteria should be specified prior to all sterilisation processes. High bioburden acceptance criteria should not be justified by the capacity of the sterilisation process or any bioburden reducing step before sterilisation. Acceptance criteria for bioburden are discussed under the relevant sub-sections of 4.1 below.
所有灭菌工艺之前的生物负荷控制标准必须明确。设定高生物负荷限度标准不应使用灭菌工艺的能力,或任何灭菌之前生物负荷减除步骤来进行论证。生物负荷的可接受标准在4.1章节相关部分进行讨论。
【海螺研习社解读】
这个思路是值得我们高度重视的,简单的理论论证不能取代实际的标准和检测结果数据,生物负荷是非常关键的指标,对于无菌工艺而言,也意味着实际风险的大小,所以不要用“评估”的方法来取代实际的测试,一句话,还是用数据说话比较靠谱。
The levels of bacterial endotoxins in the finished product can be impacted by the bioburden and bacterial endotoxins in the components (i.e. active substance, excipients and containers), and by microbiological contaminants introduced during manufacture. To ensure an acceptable level of bacterial endotoxins in the finished product, the level of microbiological contaminants of the components should be minimal. Acceptance criteria for bioburden and, where relevant, bacterial endotoxins in components and bulk solutions should be specified.
制剂成品组分(活性物质、辅料和内包材)中的生物负荷和细菌内毒素水平,以及那些在生产过程中引入的微生物污染都可能会对制剂成品细菌内毒素水平产生影响。为了确保制剂成品中的细菌内毒素水平可以接受,制剂组分中的微生物水平应尽可能降低。如果有关联,应提供制剂组分和散装溶液中内毒素和生物负荷的质量标准限度。
【海螺研习社解读】
对于大部分的企业来说,目前都知道要有相关的内毒素和生物负荷的标准限度,但是关键是这个标准如何设置,是否简单的符合USP的通则要求就可以接受?在这里,良好的商业合作流程,这个标准应该是制剂客户给自己的上游企业提出来,因为只有制剂客户自己知道自己后续有哪些工艺,最差的内毒素和生物负荷是自己可以承受的。而对于那些计划做欧美市场的原料药企业来说,如果自己的注册部门或是技术部门有能力知道后续制剂产品的规格,制剂的通用工艺水平,那么主动的提出自己的“绝对安全”的标准,绝对是加分项。对于包材辅料企业比较复杂,因为大量的包材辅料的质量人员对于制剂客户的“限度要求”实际不具备这样的知识,也不愿意花时间去了解这些信息要求,所以对制剂客户的采购来说,这是一个工作挑战了,对于制剂客户的QA来说,现场审计辅料和包材供应商的时候,供应商现场的内毒素和生物负荷控制能力、手段以及检测能力变成一个很重要的点。
All filters used in the manufacture of the finished product that come in contact with the finished product, or with any component (substance or intermediate product) incorporated in the finished product should be described and the information stated in Table3, section 4.1.5 should be provided in the quality dossier. The information should be in line with the requirements stated in Eudralex GMP Annex 1. For ATMPs, the Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products should be followed.
在制剂生产过程中所有和成品以及制剂组分(活性物质或中间体)接触的过滤器都必须进行描述,这些信息必须在质量文档的4.1.5章节的表格3进行阐述。相关信息必须符合Eudralex GMP附录1的要求。对于ATMPs产品,还必须遵守ATMPs产品优良制造指南。
【海螺研习社解读】
和物料接触的过滤器,用的好,是无菌工艺有效的保证;用的不合适或是用的不好,则是一个潜在的微生物污染源。这个绝对是辩证统一的一对矛盾,国内企业在产品价格敏感的背景下,往往对过滤器的实际使用、清洗、消毒、维护保样以及选型都会不同程度的忽略质量角度的考量。我们有机会会组织关于无菌过滤器相关指南和文献的讨论和解读,在这里就先行忽略了。
If a secondary container (e.g. secondary pouch for infusion bags or blisters intended to keep the outside of the container sterile) is used to provide a specific protection to the medicinal product, the packaging process should be described, including a risk assessment, since it may affect the sterility of the finished product; for example, trapping moisture between the primary and secondary containers. Information should be provided as to when the packaging step is performed (before or after sterilisation) and any aseptic techniques employed. The proposed processes should be justified from a microbiological perspective. If the use of a secondary container means additional sterilisation of the finished product is performed, this should be justified with regard to sterility assurance and any potential impact on finished product quality.
如果使用了外包容器(例如,输液袋的外袋,或者是为了保护内包装无菌性外表而使用的泡罩)来为药品提供特定保护,则应描述包装工艺,包括风险评估,因为这可能会影响最终产品的无菌性,比如在内包装和第二层包装之间形成湿汽。应要提交信息说明什么时候进行包装(灭菌之前还是之后)以及采用的是什么无菌技术。所采用的工艺应从微生物角度进行合理化解释。如果使用外包装意味着要对药品进行额外灭菌,则要对额外灭菌的无菌性保证能力进行解释,以及评估对药品质量的所有可能影响。
Documentation regarding sterilisation and aseptic processing to be included in the quality dossier is presented below. The documentation could, for practical reasons, be presented in connection with the item which is to be sterilised if a reference to the location of the documents is provided in section
3.2.P.3.3 or in Part 2 B. The documents may be provided for human products in sections 3.2.S.2 Manufacture, 3.2.P.2 Pharmaceutical development, 3.2.P.3 Manufacture, 3.2.P.4 Control of excipients, or 3.2.P.7 Container closure system, or for veterinary products in Part 2 A.4 Development pharmaceutics, Part 2 B.1 Manufacturing method, Part 2 C.1 Active substance, Part 2 C.2 Excipients or Part 2 C.3 Container closure systems. The documentation should be provided for all sites performing sterilisation or aseptic processing, regardless of whether the processes are performed in-house or outsourced.
灭菌和无菌工艺相关的文件归档在质量文档的以下章节:这些文件和3.2.P.3.3或第2 B部分列出的需要灭菌处理的组分相关联。具体文档归档在人用药质量文档的模块三第3.2.S.2原料药生产、第3.2.P.2药物研发、第3.2.P.3制剂生产、第3.2.P.4辅料控制或第3.2.P.7密封包装系统中的灭菌和无菌工艺文件,或者是兽药质量文档Part 2 A.4药物研发、Part 2 B.1生产方法描述、Part 2 C.1 原料药、Part 2 C.2 辅料或Part 2 C.3 密封包装系统. 要提供实施药品灭菌或无菌工艺的所有场所或无菌工艺相关的文件,不管是在公司内部还是外包给合同制造设施进行。
Process parameters such as processing and holding times are assessed and agreed during the evaluation of the quality dossier. These may be further reviewed during GMP inspections, which may result in changes to the registered dossier being required.
工艺时间和保存时间之类的工艺参数需要在质量文件评价过程中进行评估和达成共识。这些也会在后续的GMP检查中进一步被审核确认,这也可能会导致相关注册文件的变更。
4.1.1. Steam Sterilisation
蒸汽灭菌
All steam sterilisation processes require a minimum lethality of F0≥ 8 minutes and a minimum process hold temperature of 110 °C.
所有蒸汽灭菌工艺都要求F0≥ 8分钟,以及最低工艺保温温度为110 °C。
【海螺研习社解读】
这个就是底线要求。F0值/F值要搞清楚,就不得不了解D值,Z值,本篇没有篇幅去解释这些参数的实际含义和科学道理,只能简单粗暴的假设我们的读者是能理解这些参数的含义的。但是关于最低温度设定在110°C,是很有意思的一个设定。理论上,我们没有任何科学的理由来支持这个温度点的设定。考虑到F0值的限度要求,保温温度越接近121°C,就越可以在尽可能短的时间内实现这个目标。所以这个最低温度是越高越好的,但是随着温度的增高,实际压力也是需要增加的,理论上110°C的蒸汽压力就是1.43个大气压了,而121°C的蒸汽压力就是2.05个大气压了。而且在这个温度下计算F0值是最简单的,保温110°C八分钟,算出来的F0值也正好是8,符合F0值的最底线要求。可能正是综合考虑了安全性和时间效率两个角度,欧盟官方最后选择了这个110°C的最低温度点。
Sterilisation processes of different levels of lethality are presented in Table 1, along with the documentation to be included in the quality dossier. The processes in the table are presented with decreasing lethality when read from top to bottom, thus the first feasible process should be selected.
不同致死率的灭菌工艺参见表格1,也包括需要在质量文档涵盖的文档内容。表格中的工艺从上到下罗列了逐渐降低的致死能力,所以优选选择第一个可行的工艺参数。
For sterilisation using a reference condition of the Ph. Eur. 5.1.1 (≥121 °C, ≥15 min in all units) validation data for the sterilisation cycle is not required to be submitted in the quality dossier.
对于使用EP5.1.1参考条件(所有包装均≥121°C, ≥15 min)的灭菌,不需要在质量文档中提交灭菌循环的验证数据。
【海螺研习社解读】
这个其实是最优选择,优越到你都不需要递交数据给官方了!因为在这个条件下,即便不做任何的计算,你也直接可以发现这个条件是之前F0值=8的要求的接近两倍了,是属于典型的过度灭菌了,那么还有啥好说的了?
If used as an additional control to measure the process lethality, F0, should be stated, together with the lowest temperature measured by the temperature sensors to determine F0.
如果使用额外控制手段对工艺灭菌能力的进行监测, 则应说明该方法的F0值, 以及由温度探头测得的最低温度来确定F0值。
Steam sterilisation performed with finished product temperature below 115 °C during the holding phase is an exceptional case and should be scientifically justified and supported by additional data as described in Table 1. If temperatures below 110 °C are included (during heat-up and cool-down) in the determination of F0, this should be justified.
成品温度低于115°C保持一定时间的蒸汽灭菌是一种特殊情况, 应进行科学论证, 并有表格1所述的其他数据的支持。如果在确定F0值的数据中包括低于110°C 的温度 (在加热和冷却过程中), 应进行论证。
【海螺研习社解读】
这个规定实际约定了任何低于110°C的过程,无论其时间长短以及实际对灭菌的贡献,均不应该参与到无菌F0值的计算过程中去。
Information regarding the F0 concept and microbial reduction is provided in Ph. Eur. 5.1.5 Application of the F0 concept to steam sterilisation of aqueous preparations.
关于F0概念和微生物降低的信息见 Ph. Eur. 5.1.5 F0值在水溶性制剂蒸汽灭菌中的应用。
The bioburden limit should be in line with any pre-sterilisation bioburden reduction process capability (e.g. filtration). For aqueous solutions, the limits stated in Table 1 are acceptable for active substances and drug product formulations without further justification. Other testing regimes and limits to control bioburden at the defined level should be justified.
微生物负荷限度应与任何预灭菌工艺的微生物负荷降低能力 (如过滤) 一致。在没有特殊说的情况下,对于水溶液,表格1所述的限度对于活性物质和药物产品制剂是可以接受的。其他用来控制微生物负荷处于既定水平的测试方法和限度应进行论证。
Moist heat processes with an F0<8 min may be suitable as a post-aseptic processing terminal heat treatment for formulations that cannot withstand a complete terminal sterilisation cycle. Such processes may further ensure a SAL of sterile filtered (or otherwise sterilised) bulk components, which have been aseptically filled. Post-aseptic processing terminal heat treatments are also presented in Table 1.
F0值< 8分钟的湿热工艺可作为无菌处理后的终端热处理, 适用于无法承受完整的终端灭菌程序的制剂。此类工艺可进一步确保经无菌过滤(或以其他方式灭菌) 后进行无菌分装的大批量组分的SAL。无菌工艺后终端热处理也见表格1。
It is emphasised that this additional post-aseptic processing terminal heat treatment should not compensate for poor aseptic manufacturing practice. The same requirements for the aseptic part of the process apply as for finished products manufactured without such an additional post-aseptic processing terminal heat treatment.
需要强调的是, 这种额外的无菌工艺后进行终端热处理的工艺不应用来作为不良无菌生产操作的补偿。这与没有额外无菌加工终端热处理在无菌工艺部分的要求是一样的。
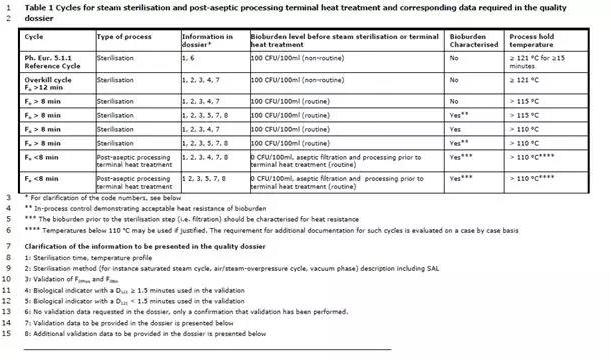
表格1 蒸汽灭菌循环和无菌后处理终端热处理工艺以及相关的质量文档数据要求
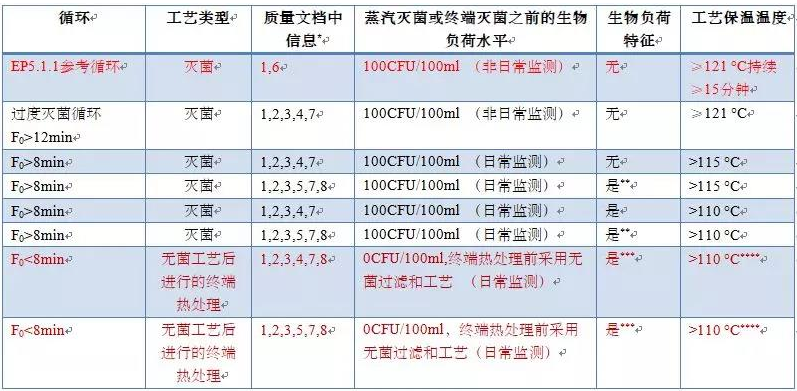
*代码含义参见以下的解释
**中控证明负载微生物的耐热性是可接受的
***除菌步骤(例如过滤)前的负载微生物应进行耐热性确认
****110C以下的温度需要进行合理化解释,这些情况下需要额外的补充资料。
质量文档中需要的文件信息:
1. 灭菌时间,过程温度档案
2. 灭菌方法(例如饱和蒸汽周期,空气/蒸汽过饱和压力循环,真空)描述,包括SAL
3. F0phys和F0iso验证
4. 验证中使用的生物指示剂(D121≥1.5分钟)
5. 验证中使用的生物指示剂(D121<1.5分钟)
6. 文档中不需要验证文档,只需要确认验证做了
7. 质量文档需要递交验证数据
8. 质量文档需要额外的验证数据支持
Validation data to be provided in the quality dossier for all steam sterilisation processes that do not fulfill the requirements of Ph. Eur. 5.1.1 standard process (required information 7 in Table 1):
对于不符合 Ph. Eur. 标准工艺要求的所有蒸汽灭菌工艺, 需在质量档案中提供的验证数据 (表格1中所需信息 7):
Load mapping of the chamber and load mapping distribution of the items in the chamber (including the slowest to heat locations); summary or confirmation of performance.
腔体的装载布局和腔体中物品的装载布局图(包括加热最慢的位置); 性能确认总结。
Physical and biological cycle effect confirmation summary of at least three sterilisation runs demonstrating an SAL ≤10-6, as described in Ph. Eur. 5.1.1 ensuring:
至少三次灭菌程序运行的物理和生物效果确认总结, 显示SAL≤10-6, 如 ph. Eur第5.1.1章节的要求:
Demonstration that the sterilisation load in the steriliser chamber achieves the specified cycle parameters, including time, temperature, pressure and F0, if applicable;
显示灭菌腔室中的灭菌装载达到了规定的循环参数, 包括时间、温度、压力和F0值 (如适用);
Acceptable temperature differences between temperature sensors in the load;
装载中温度探头之间温差可接受;
Acceptable F0 variability within the load;
装载中可接受的 F0值波动;
Relationship between physical and biological validation.
物理和生物学验证之间的关系。
For the biological validation, a biological indicator as described in Ph. Eur. chapter 5.1.2 Biological indicators and related microbial preparations used in the manufacture of sterile products with a D121-value of ≥1.5 minutes should be used.
对于生物学验证, 应使用Ph.Eur. 5.1.2章节 “用于无菌产品生产的生物指示剂和相关微生物制品”中规定的D121℃值≥1.5分钟的生物指示剂。
The SAL should be determined, its microbiological basis should be justified and details of calculations provided in the quality dossier. Preferably it should be calculated from the maximum bioburden per container and the D-value of the biological indicator used in the validation.
应确定SAL,其微生物基础应合理,并在质量档案中提供计算细节。最好是根据每个容器的最大生物负荷和验证中使用的生物指示剂的D值来进行计算。
Additional validation data to be provided in the quality dossier for low energy steam processes or where a bio-indicator with a D121-value of <1.5 minutes is used in the validation of the sterilisation process (required information 8 in Table 1):
在低杀灭率蒸汽灭菌工艺的质量档案中提供的补充验证数据,或在灭菌过程的验证中使用D121值<1.5分钟的生物指示剂的情况下需提供的补充验证数据(表格1中所需信息 8):
The following additional data should be provided:
应提供以下补充数据:
A justification for the start point of the sterilisation phase, that is the temperature when the temperature sensors record the F0 from the start to end of the process;
论证灭菌段的起点,即灭菌开始至结束温度探头用于记录F0的温度;
Biological indicators with suitable resistance at the actual temperature range as described in Ph. Eur. 5.1.2 should be included in the validation to demonstrate sensitivity to the process.
验证应使用在 Ph.Eur.第5.1.2章中所述的实际温度范围内具有适当耐热性的生物指示剂,以证明对工艺的敏感性。
More detailed validation data is requested to ensure that the proposed sterilisation process is suitable for low temperature processes and for processes using biological indicators of low heat resistance because:
为了确保使用低温工艺和使用低耐热性生物指示剂的灭菌工艺是适当的,需要更详细的验证数据, 因为:
The change in lethal effect in relation to the process temperature may not be log linear at lower sterilisation temperatures.
在较低的灭菌温度下, 杀灭效果与工艺温度的关系可能不是呈对数线性的。
The SAL demonstrated in the validation of a sterilisation process is dependent on the heat resistance of the biological indicator used in the validation of the process. When a biological indicator of low D-value is used in the validation of the sterilisation process, the SAL demonstrated becomes numerically higher, but does not provide as high a safety margin as where a more resistant biological indicator is used. The SAL should always be established in relation to a D-value that is higher than that of the normal bioburden at routine production.
在灭菌工艺的验证中显示的SAL取决于在验证过程中使用的生物指示剂的耐热性。当在灭菌过程的验证中使用低D值的生物指示剂时, 所显示的SAL会变得更高,但不能提供与使用更耐热的生物指示剂那样高的安全系数。SAL应该基于比日常生产中正常生物负荷高的D值来建立。
【海螺研习社解读】
这个非常全面,没有更多需要解释的了,唯一要做的就是把它打印出来贴在自己办公室的墙上吧!对应的科学解释,后面两段话的解释也很具体了。如果还是看不懂的朋友,要去先学习下D值,SAL,F值,Z值这些基本概念了。
唯一需要提醒的是,在应用本指南的要求的时候,不能生搬硬套,实际工艺日常生产中的生物负荷,实际的可能的菌种信息,工厂都应该尽可能多的收集,然后基于这些实际的数据和实际的情况,来科学的选择适合自己的生物指示剂,来进行科学的这些灭菌工艺值的计算,然后合理的去解释给监管当局。整齐划一的试图用一个标准,一种方法,一种生物制试剂去解决自己多个产品多条生产线的所有问题,并不是好的“质量文化”和科学的质量态度,往往会后日后的运行管理带来潜在的不可预期的风险。
4.1.2. Dry heat sterilization
干热灭菌
Time and temperature of the sterilisation cycle and a bioburden limit should always be stated.
应说明灭菌周期的时间和温度以及生物负荷限度。
For sterilisation using a reference condition of the Ph. Eur. 5.1.1 (a minimum of 160 °C for at least 2 h), the validation data for the sterilisation cycle is not required to be submitted in the quality dossier. For sterilisation cycles with time and/or temperature lower than the reference conditions of the Ph. Eur., physical and biological validation of the sterilisation cycle should be provided to demonstrate an SAL of ≤10-6, as described in Ph. Eur. 5.1.1. The SAL of such a sterilisation process should be calculated from the maximum bioburden per container.
对于使用 Ph. Eur. 5.1.1 的参考条件 (≥160℃至少2小时 ) 进行灭菌, 灭菌周期的验证数据不需要在质量档案中提交。对于时间和温度低于药典参考条件的灭菌周期,应提供灭菌周期的物理和生物验证, 以证明 SAL≤10-6。这种灭菌工艺的SAL应根据每个容器的最大生物负荷计算。
Where required, sufficient validation data should be submitted to demonstrate that an SAL of ≤10-6 is obtained for all containers. The data submitted should include at least, but is not limited to:
必要时,应提交足够的验证数据, 以证明所有容器都能获得≤10-6 的 SAL。提交的数据至少应包括但不限于:
Load mapping of the chamber and load mapping distribution of the items in the chamber (including the slowest to heat locations) – summary or confirmation of performance;
腔体的装载布局和腔体中物品的装载布局图(包括加热最慢的位置)- 性能确认总结;
Physical and biological cycle effect confirmation summary of at least three sterilisation runs ensuring:
至少3次灭菌周期的物理和生物性能确认总结, 以确保:
Demonstration that the sterilisation load in the steriliser chamber achieves the specified cycle parameters, including time, temperature, and, lethality;
证实灭菌器腔体中的装载达到了规定的循环参数, 包括时间、温度、致死率等;
Acceptable temperature differences between temperature sensors in the load;
负载中温度探头之间可接受的温度差异;
Acceptable lethality variability within the load;
负载内杀灭率可接受的波动;
Relationship between physical and biological validation.
物理和生物学验证之间的关系。
For the biological validation, a biological indicator as described in Ph. Eur. chapter 5.1.2 should be used.
对于生物学验证, 应使用Ph. Eur. 5.1.2章节所述的生物指示剂。
A maximum bioburden limit of 100 CFU/100 g or 100 CFU/100 ml would be acceptable for parenteral finished product formulations without further justification. For active substances and finished products that are not used for parenteral administration, a maximum total bioburden limit of 10 CFU/g or 10 CFU/ml is acceptable without further risk based justification. Other testing regimes and limits to control bioburden at the defined level should be justified. A justified bioburden limit should also be established for empty containers.
对于注射用制剂处方, 最大微生物负荷限制为 100CFU/100g 或 100 CFU/100 毫升是可以接受的,不需要进一步论述。对于非注射用制剂处方和活性物质, 最大微生物负荷限度为 10 CFU/g或 10 CFU/ml 是可以接受的,不需要进一步的风险论证。其他用以控制微生物负荷在规定水平内的方法和限度应进行论证。对于任何空的容器,也应建立合理的生物负荷限度。
Dry heat at temperatures of greater than 220 °C for a validated time is frequently used for both sterilisation and depyrogenation of glassware and other heat-resistant container materials e.g. aluminium crimps. In this case, demonstration of a 3 log reduction in heat-resistant endotoxins can be used as validation criteria.
在经过验证的时间内,温度高于220°C的干热灭菌经常被用于玻璃器皿和其他耐热容器材料,比如压制铝的灭菌和去热原。在这种情况下, 证明能够将耐热内毒素下降3个log等级作为验证的标准。
【海螺研习社解读】
对于注射用制剂、非注射用制剂,和原料药的微生物负荷的限度要求基本上都是行业内普遍接受的标准。虽然这个指南说不需要进行进一步的风险评估和合理化解释,但是说实话,考虑到产品的差异性以及微生物种类的差异性,这样的生物负荷的限度要求是值得推敲的。测试样品的数量规定也是一个很大的问题。这些都是实际操作中可能带来争议的地方,也会给样品的代表性带来质疑的空间。对于负责的企业而言,最好让自己的微生物专家,结合产品的实际和生产制造的实际,潜在微生物种类的实际,进行周全的风险评价,然后制订符合自己要求和实际的标准还是我们最佳的选择。
干热温度设定的底线是220°C, 这个温度点的设定基础是什么?指南没有解释,更加常见的还有250°C这个点。就内毒素而言,干热破坏的温度点250°C或许更加常见。当然时间的差异和温度的高低直接相关联,所以指南要求对时间进行验证。
4.1.3. Ionization radiation sterilization
电离辐射灭菌
For this method of sterilisation, the reference absorbed dose is ≥25 kGy. Other doses may be used to achieve an SAL ≤10−6, if justified and validated.
对于这种灭菌方法, 参考吸收剂量为≥25 kGy。如经论证和验证,也可以使用其他剂量以达到SAL≤10-6。
Data as requested in Note for Guidance “The use of Ionization Radiation in the Manufacture for Medicinal Products” and in compliance with Ph. Eur. chapter 5.1.1 should be provided. Relevant guidance in establishing the radiation dose other than 25 kGy is available in ISO standard 11137.
应提供“药品生产中电离辐射使用指南”中要求的数据, 并符合Ph. Eur 5.1.1 章节的要求。ISO 11137标准提供了确定 25 kGy 以外的辐射剂量的相关指导。
Where any requirements in ISO 11137 are in contradiction to requirements stated in any Note for Guidance issued by the EMA or Ph. Eur. monograph, the requirements of the Ph. Eur. and the Note for guidance apply.
如果 ISO 11137标准中的任何要求与 EMA 或 Ph. Eur. 专论发布的任何指导说明中的要求相抵触, 则应使用 Ph. Eur. 和医药指南的要求。
【海螺研习社解读】
这一规定其实表明了EMA对于ISO相关标准的态度。对于采用电离辐射灭菌的很多企业实际采用的是合同制造企业进行的操作,这些合同组织很多时候不会单纯的服务医药行业,所以更多的会在体系中遵循ISO标准,对于医药行业的QA在进行这类合同组织审计的时候,不能因为对方声称符合ISO标准就对其放松要求,要仔细核对细节,确保ISO标准和要求和现有的EMA医药指南和EP药典的相关规定相冲突。
4.1.4. Gas sterilization
气体灭菌
4.1.4.1 General considerations
通则
Generally, gas sterilisation is only acceptable if no other method of sterilisation is possible. Gas sterilisation provides sterilisation of the surface of materials. It is mainly employed for sterilising packaging materials and equipment, and has therefore only been included in the decision tree for containers. To ensure adequate sterility, sufficient penetration by gas and moisture is essential. This should be followed by a purging process to ensure that any residues of gas or related transformation by-products are below concentrations that could give rise to toxic effects during use of the finished product. The effectiveness of the purging process should be demonstrated.
一般来说, 只有在没有其他灭菌方法的情况下, 气体灭菌才是可以接受的。气体灭菌可对材料表面进行灭菌。它主要用于对包装材料和设备进行灭菌, 因此只包括在容器的决策树中。为了确保足够的无菌性, 确保气体和水分能够充分渗透是非常重要的。气体灭菌结束后应进行清除过程以确保灭菌气体或其相关的转化形成的副产品的任何残留物低于可能提高成品使用过程中的毒性影响的浓度。应证明清除过程的有效性。
Gas sterilisation of porous compounds, such as dry powders, is not acceptable unless other methods of sterilisation are not feasible and its use is scientifically justified. Prior to the gas sterilisation, the active substance or excipient should be sterile filtered and crystallised under aseptic conditions to minimise bioburden and entrapment of micro-organisms within the crystals. Convincing evidence should be provided demonstrating that the material to be sterilised is not susceptible to compression preventing gas and moisture penetration during sterilisation.
对多孔化合物 (如干粉) 进行气体灭菌是不可接受的, 除非其他灭菌方法不可行且使用该方法经科学论证。在气体灭菌之前, 活性物质或辅料应在无菌条件下进行除菌过滤和结晶, 以最大限度地减少微生物在晶体中的吸收和包裹。应提供令人信服的证据, 证明待灭菌的材料在灭菌过程中不易挤压,防止气体和水分渗透。
A description of the apparatus, quantitative data on gas(es) to be used, the bioburden prior to sterilisation, the time of exposure to the gas, the temperature and humidity prior to and during each step of the sterilisation cycle, and, if applicable, the conditions for the removal of any toxic gas residues should be provided. Humidity used for the preconditioning and/or conditioning of the material to be sterilised shall be generated by clean steam. These conditions should be monitored by appropriate in-process controls with justified acceptance criteria. The process should be developed and validated in compliance with Ph. Eur. 5.1.1 and 5.1.2. A risk assessment with regards to residual toxic impurities should be conducted and a control strategy should be provided where applicable. The requirements should be in accordance with the requirements of ICH M7 “Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk”. Even if the relevant product is outside the scope of that guideline, its limits for highly toxic impurities could be applied.
应提供设备描述、要使用的气体的定量数据、灭菌前的生物负荷、接触气体的时间、灭菌周期每一步开始前和期间的温度和湿度, 以及,适当时,清除任何有毒气体残留的条件。用于对待灭菌材料进行预处理和/或调节的湿度应通过清洁蒸汽产生。这些条件应通过适当的过程控制和合理的接受标准进行监测。应按照 Ph.Eur. 5.1.1 和5.1.2 章节制定和验证这一程序。应对残留有毒杂质进行风险评估, 并酌情提供控制战略。这些要求应符合 ICH M7指南 “评估和控制药品中的 DNA 反应性 (诱变) 杂质以限制潜在致癌风险”。即使相关产品可能不适用于该指南的范围, 该指南中对剧毒杂质的限度也是可以使用的。
Results of the process validation should demonstrate an SAL of ≤10-6.
工艺验证的结果应证明SAL ≤10-6。
The effectiveness of the process should be routinely checked for every batch confirming that the process parameters and biological indicators are all within their acceptance criteria and by sterility testing. Parametric release is not acceptable for gas sterilisation (according to Ph. Eur. chapter 5.1.1).
日常应通过确认工艺参数和生物指示剂都在其接受标准范围内和无菌测试来检查每一批工艺的有效性。参数放行对于气体灭菌是不可接受的 (参考Ph. Eur. 5.1.1 章节)。
【海螺研习社解读】
采用气体对产品进行灭菌永远是最后的选择,不得以采用的选择,因为气体和灭菌目标物往往出于不同的状态,而且比蒸汽灭菌更加复杂的是因为温度的限定往往杀菌作用是和接触时间和接触浓度相关的,而接触浓度会带来潜在的灭菌气体和目标物之间的化学反应,这些要素都让灭菌过程更加复杂,也给产品引入新的杂质机会。杀菌气体,由于其固有的杀菌特性决定了其对于正常人体细胞的毒性,所以其残留是不被允许的,给消毒后的残留检测标准和检测方法有带入了新的问题。这些都是气体灭菌不被首先考虑的科学基础。
在实际操作中,当气体灭菌的决策被最后采纳后,我们强烈的建议公司要耐心的进行各项基础的实验去收集尽可能多的信息和数据,然后根据这些数据和信息来制订科学完善的灭菌工艺流程和相关的参数,并确保操作人员严格按照相关的流程和操作细节进行生产操作。如果你是审计人员,工厂存在这个类型的操作的时候,不得不高度重视我们在这里提到的这些环节了。
4.1.4.2 Ethylene oxide sterilization
环氧乙烷灭菌
Ethylene oxide (ETO) sterilisation processes should be developed and validated in compliance with Ph. Eur. 5.1.1 and 5.1.2. Relevant guidance in establishing the sterilisation process cycle parameters and validation is available in ISO standard 11135.
应按照 Ph. Eur 5.1.1 和5.1.2 章节开发和验证环氧乙烷 (ETO) 灭菌工艺。ISO 11135标准提供了在确定灭菌工艺循环参数和验证方面的相关指导。
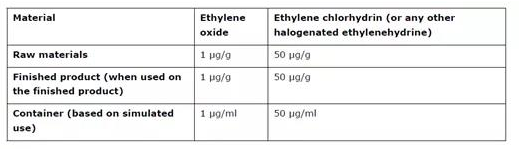
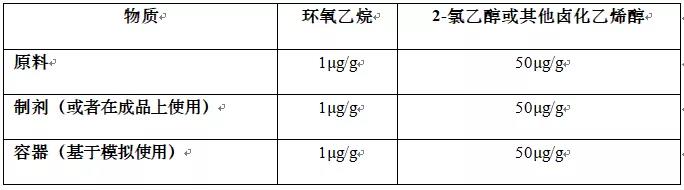
ETO is a gas which is highly toxic. ETO sterilisation is generally only acceptable if no other method of sterilisation is possible. The risk assessment should consider the residual known genotoxic impurities (such as ETO and halogenated ethylenehydrines). This should be evaluated in accordance with the requirements of ICH M7 “Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk”, unless the relevant product is outside the scope of that guideline. For products outside the scope of ICH M7, the applicant should apply limits for highly toxic impurities in accordance with ICH M7, or the acceptance criteria stated in Table 2, whichever is most appropriate.
ETO 是一种高毒性气体。ETO 灭菌通常只有在没有其他灭菌方法的情况下才可接受。风险评估应考虑已知的残留遗传毒性杂质 (如 ETO 和卤化乙二氢)。应根据 ICH M7 指南"评估和控制药品中的 DNA 反应性 (诱变) 杂质以限制潜在致癌风险" 的要求对此进行评估, 除非相关产品不在该指南的范围之内。对于 ICH M7 范围以外的产品, 申请人应根据 ICH M7指南 或表格2中规定的接受标准对剧毒杂质实施限制, 以最适当者为准。
For empty containers intended to be filled with aqueous products, (e.g. prefilled syringes), the need to justify the use of ETO in the sterilisation of the container prior to filling can be waived, as the degradation kinetics of ETO in an aqueous medium have been sufficiently demonstrated. However, the levels of toxic residues (ETO and halogenated ethylenehydrines) in the finished product need to fulfil the requirements of ICH M7, or the limits stated in Table 2 below, as applicable.
对于打算装满水溶性性产品的空容器 (例如预填充注射器), 可以免除对灌装前使用ETO对容器灭菌的论证,因为ETO在水介质中的降解动力学已经充分证明。但是, 成品中毒性残留物 (ETO 和卤化乙基氢) 的含量需要满足 ICH M7 的要求,或者适当时,满足下文表格2所述的限度。
Table 2 Limits for toxic gas residues from Ethylene Oxide sterilisation where the ICH M7 limits do not apply
文章来源:海螺研习社
本网站刊载的所有内容,包括文字、图片、音频、视频、软件等,如非标注为“原创”,则相关版权归原作者所有,如原作者不愿意在本网站刊登相关内容,请及时通知本站,我们将第一时间予以删除。